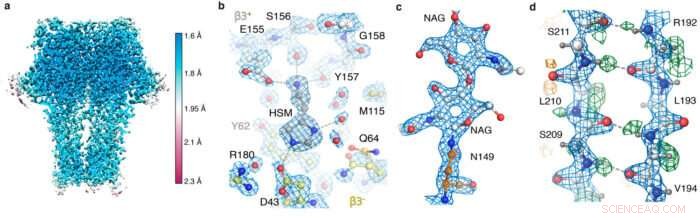
GABA A receptorkarta ögonblicksbilder. (a) Lokal resolution. (b) agonistfickan som visar histaminkoordination och vattenmolekyler; (c) N-kopplad glykan; (d) vätebindningsnätverk som avslöjas av skillnadskartan (gröna toppar).
Att titta på det exakta tredimensionella arrangemanget av atomer i ett protein hjälper oss att förstå hur det kan utföra sina funktioner. Även om elektronkryo-mikroskopi (kryo-EM) har utvecklats snabbt som en viktig strukturbiologisk teknik under de senaste åren, Röntgenkristallografi hade varit den enda tekniken som kunde visualisera enskilda atomer. Radu Aricescus och Sjors Scheres grupper vid MRC Laboratory of Molecular Biology, i samarbete med forskare vid Thermo Fisher Scientific och på andra håll, har nu för första gången kunnat lösa upp enskilda proteinatomer i en tredimensionell kryo-EM-bild.
Detta samarbete startade i början av 2019 när Radu och Abhay Kotecha, en forskare vid Thermo Fisher Scientific, ville testa ny cryo-EM-hårdvara på ett litet membranproteinprov. GABAA-receptorer, ett fokus för Radus forskning i över ett decennium, valdes för att den högsta möjliga upplösningen med bästa tillgängliga teknik verkade ha nått en gräns vid cirka 2,5 Ångströms (Å), men högre upplösning behövdes helt klart för bättre läkemedelsdesign.
Vad är atomupplösning?
Upplösning brukar redovisas i Ångströms, en längdenhet som är en tiomiljarddels meter eller 0,1 nanometer, och hänvisar till det minsta avståndet mellan vilket två objekt kan ses vara åtskilda.
Längden på en typisk kol-kolbindning är 1,5 Å; andra bindningar i proteiner är lite kortare. Således, när upplösningen går ner till 1,2 Å, det blir möjligt att se enskilda atomer i ett protein, uppnå verklig atomupplösning.
Medan jag testade ny hårdvaruutveckling som inkluderade en elektronkälla för kallfältsemissionspistoler, ett nytt energifilter, och en ny kamera, teamet fick också utveckla nya bearbetningsstrategier. Algoritmer för korrigering av optiska aberrationer som tidigare utvecklats av Jasenko Zivanov i Sjors grupp, samt en algoritm föreslagen av Chris Russo och Richard Henderson, spelade en avgörande roll för att pressa ut mest information ur bilderna.
Efter att ha tagit emot bilder insamlade på den nya mikroskophårdvaran av Abhay Kotecha på Thermo Fisher Scientific i Eindhoven, Nederländerna, Takanori Nakane, postdoc i Sjors grupp, utvecklat ett optimalt arbetsflöde i RELION och Andrija Sente, tillsammans med andra medlemmar i Radus grupp, använde detta arbetsflöde för att bearbeta GABAA-receptorbilder, samtidigt som resultaten återkopplas för att snabbt optimera mikroskopinställningarna. En ny, Datalagringssystem med hög kapacitet utvecklat av Jake Grimmett och Toby Darling i LMB:s Scientific Computing-team erbjöd avgörande stöd för att hantera de cirka hundra terabyte av data som genererades. Denna ihållande teaminsats ledde till en oöverträffad 1,7 Å-upplösning GABAA-receptorstruktur.
Detta var den bästa rapporterade upplösningen som uppnåddes med användning av cryo-EM för något annat proteinprov än för proteinet apoferritin. Apoferritin används ofta som ett riktmärke för cryo-EM, eftersom dess molekylära stabilitet och 24-faldiga symmetri tillåter högupplösta rekonstruktioner från relativt få partiklar.
Genom att använda den nya hårdvaran och bearbetningsstrategierna, teamet kunde erhålla en 1,22 Å upplösning apoferritinstruktur, slog det tidigare rekordet på 1,53 Å för att vara den högsta upplösningen av enpartikel-kryo-EM-struktur som hittills erhållits. Mest imponerande, denna upplösning möjliggjorde visualisering av individuella väteatomer, även på vattenmolekyler inuti proteinstrukturen. Visualiseringen av vätebindande nätverk inuti proteinstrukturer och i läkemedelsbindningsfickor gör att forskare bättre kan förstå hur de fungerar.
Detta arbete representerar brytandet av en nyckelbarriär för cryo-EM som en strukturell biologiteknik och den nya teknologin, datainsamling, och bearbetningsstrategier kommer att utöka antalet proteiner vars strukturer kan lösas till hög upplösning. Dessa rekonstruktioner med högre upplösning kommer att möjliggöra en bättre förståelse av hur proteiner fungerar och underlätta design av mer specifika läkemedel som kan påverka behandlingar för ett stort antal sjukdomar.