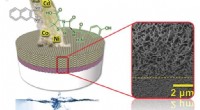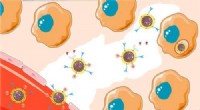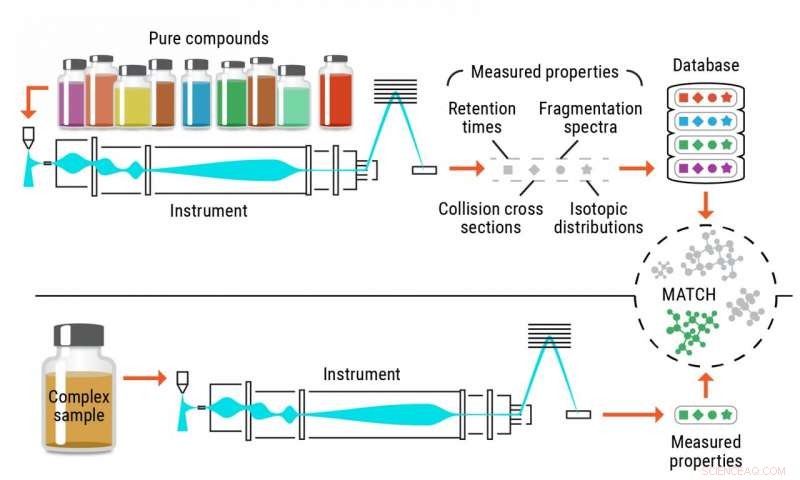
Illustration av den konventionella identifieringsprocessen av metaboliter. Kredit:Pacific Northwest National Laboratory
Noggrann identifiering av metaboliter, och andra små kemikalier, i biologiska och miljömässiga prover har historiskt kommit till korta vid användning av traditionella metoder. Konventionell taktik bygger på rena referensföreningar, kallas standarder, att känna igen samma molekyler i komplexa prover. Dessa tillvägagångssätt begränsas av tillgången på de rena kemikalier som används som standarder.
"Vi ville verkligen kringgå det nuvarande paradigmet om hur ett metabolomikexperiment genomförs och hur molekyler säkert identifieras, sa Tom Metz, biomedicinsk forskare vid Pacific Northwest National Laboratory (PNNL) och chef för Pacific Northwest Advanced Compound Identification Core.
Ett problem med den nuvarande metoden är att det bara finns så många rena föreningar som forskare kan köpa från leverantörer; de flesta leverantörer har tillgång till cirka 3, 000–4, 000 föreningar.
"Om du tänker på vad som förutspås inträffa i naturen, du tittar på> 1030 föreningar eller fler som kan vara möjliga, " sa Metz. "Så, när du jämför de få tusen standardkemikalier du har tillgång till med det stora antalet potentiella föreningar, du är inte ens nära."
Standardfri identifieringsmetod
För att lösa detta problem utvecklade Metz och hans team på PNNL ett tillvägagångssätt – standardfri metabolomik – med vilken de beräknar eller förutsäger information om flera egenskaper för molekyler av intresse för att generera omfattande referensbibliotek och sedan matcha experimentella data som innehåller samma egenskaper till dessa bibliotek, möjliggör identifiering av förening.
Genom att använda detta nya tillvägagångssätt, forskare skickar kemiska strukturer genom maskininlärning eller kvantkemiprogram för att exakt förutsäga metaboliternas experimentella egenskaper.
"Om vi är tillräckligt exakta i dessa förutsägelser så skulle vi teoretiskt sett aldrig behöva analysera en ren förening igen, " sa Metz. "Denna samling verktyg kommer att förändra det nuvarande paradigmet inom metabolomik, och inom en snar framtid kommer det att finnas några riktigt bra applikationer för att visa forskarvärlden fördelarna med detta nya tillvägagångssätt."
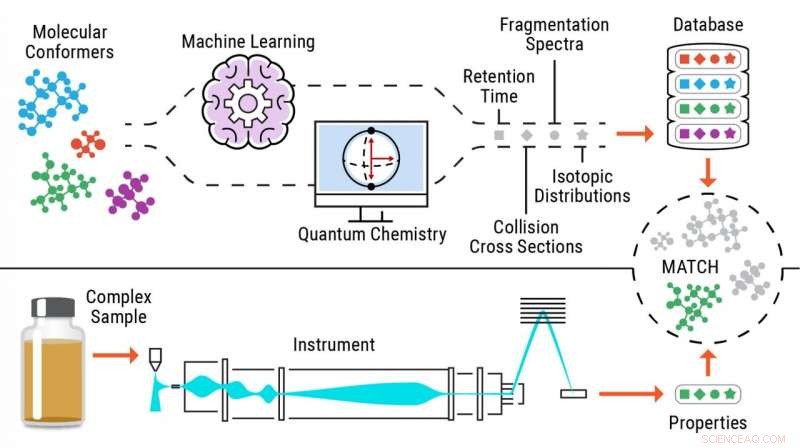
Illustration av standardfri identifieringsprocess av metaboliter. Kredit:Pacific Northwest National Laboratory
Genom att inte behöva förlita sig på data från analyser av rena standarder för att identifiera små molekyler, det standardfria tillvägagångssättet möjliggör identifiering av upp till 90 procent fler kemikalier i prover och gör dessa beräkningsverktyg mycket användbara inom flera applikationsområden, inklusive upptäckt av nya läkemedel, kemisk kriminalteknik, och miljö- och biomedicinsk forskning.
"Till exempel, i ny läkemedelsdesign skulle en användare kunna säga, "Jag har ett visst antal egenskaper med dessa vissa droger, men de råkar vara giftiga. Kan vi förutsäga en förening som skulle ha liknande egenskaper men kanske inte är giftig?'" sa Metz. "Om rätt träningsdata kunde ges till DarkChem-programmet, DarkChem kunde sedan utföra den förutsägelsen."
Anpassningsbar programsvit
Den nya metoden för standardfri metabolomikidentifiering använder fyra nyckelverktyg för att skapa omfattande, i kiselhärledda metabolitreferensbibliotek, och för att extrahera och matcha experimentella data för att ge sammansättningsidentifikationer:
Verktygen har designats för att fungera tillsammans, men de kan också användas separat. Forskare kan skräddarsy de olika applikationerna utifrån kundens behov eller forskningsområden, skapa ett helt modulärt tillvägagångssätt.
Främja ett forskningsfält
Just nu, inom metabolomics community, alla forskare identifierar samma uppsättning molekyler i varje prov. Anledningen till det är att de alla har samma rena föreningar som de köpte för att bygga ut sina referensbibliotek.
"Vår vision är att genom att använda det standardfria tillvägagångssättet kommer du aldrig att begränsas av vidden av små molekyler som kan identifieras i ett prov, " sade Metz. "Det är verkligen en spelförändring för metabolomics. Och det är väldigt spännande att se vad nästa år eller så har i beredskap för detta."