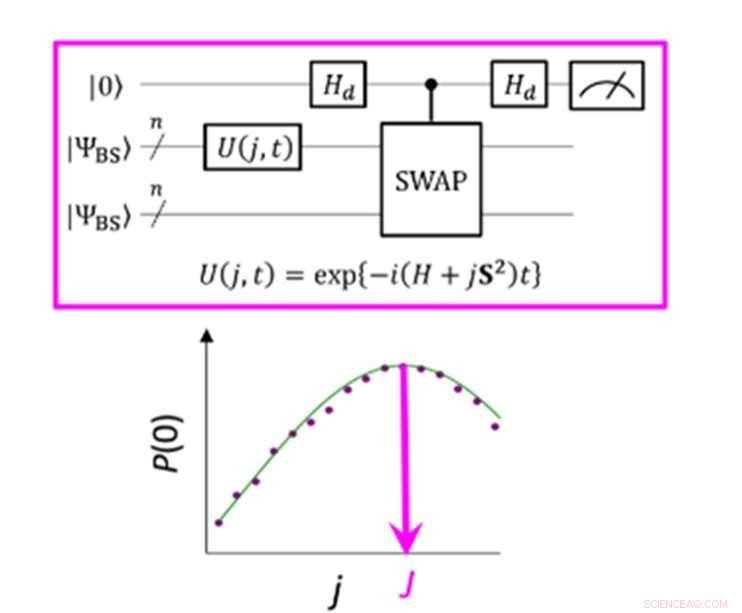
En kvantkrets som möjliggör maximal sannolikhet för P (0) vid mätning av parametern J. Kredit:K. Sugisaki, K. Sato och T. Takui
Forskare vid Osaka City University använder kvantöverlagringsstater och Bayesiansk slutsats för att skapa en kvantalgoritm, lätt körbar på kvantdatorer, som exakt och direkt beräknar energiskillnader mellan den elektroniska marken och exciterade spinntillstånd för molekylära system under polynomtid.
Att förstå hur den naturliga världen fungerar gör att vi kan efterlikna den till gagn för mänskligheten. Tänk på hur mycket vi litar på batterier. Kärnan är att förstå molekylära strukturer och beteendet hos elektroner inom dem. Att beräkna energiskillnaderna mellan en molekyls elektroniska mark och upphetsade spinntillstånd hjälper oss att förstå hur vi bättre kan använda den molekylen i en mängd olika kemikalier, biomedicinska och industriella tillämpningar. Vi har gjort stora framsteg i molekyler med slutna skalsystem, där elektroner är ihopkopplade och stabila. Open-shell-system, å andra sidan, är mindre stabila och deras underliggande elektroniska beteende är komplext, och därmed svårare att förstå. De har oparade elektroner i sitt marktillstånd, som gör att deras energi varierar på grund av elektronspinnens inneboende natur, och gör det svårt att mäta, särskilt när molekylerna ökar i storlek och komplexitet. Även om sådana molekyler är rikliga i naturen, det saknas algoritmer som kan hantera denna komplexitet. Ett hinder har handlat om det som kallas den exponentiella explosionen av beräkningstid. Att använda en konventionell dator för att beräkna hur oparade snurr påverkar energin i en öppen skalmolekyl skulle ta hundratals miljoner år, tid människor inte har.
Kvantdatorer är under utveckling för att minska detta till det som kallas "polynomtid". Dock, processen som forskare har använt för att beräkna energiskillnaderna för öppna skalmolekyler har i huvudsak varit desamma för både konventionella och kvantdatorer. Detta hämmar den praktiska användningen av kvantberäkning i kemiska och industriella tillämpningar.
"Tillvägagångssätt som åberopar sanna kvantalgoritmer hjälper oss att behandla öppna skalsystem mycket mer effektivt än genom att använda klassiska datorer, "uppger Kenji Sugisaki och Takeji Takui från Osaka City University. Med sina kollegor, de utvecklade en kvantalgoritm körbar på kvantdatorer, som kan, för första gången, exakt beräkna energiskillnader mellan den elektroniska marken och spända tillstånd i öppna skalmolekylära system. Deras resultat publicerades i tidskriften Kemisk vetenskap den 24 december 2020.
Energiskillnaden mellan molekylära spinntillstånd kännetecknas av värdet på utbytesinteraktionsparametern J. Konventionella kvantalgoritmer har exakt kunnat beräkna energier för slutna skalmolekyler "men de har inte kunnat hantera system med en stark multi-konfiguration karaktär, "uppger gruppen. Fram till nu har forskare har antagit att för att erhålla parametern J måste man först beräkna den totala energin för varje spinntillstånd. I öppna skalmolekyler är detta svårt eftersom den totala energin för varje spinntillstånd varierar kraftigt när molekylen förändras i aktivitet och storlek. Dock, "Energiskillnaden i sig är inte mycket beroende av systemstorleken, "konstaterar forskargruppen. Detta fick dem att skapa en algoritm med beräkningar som fokuserade på snurrskillnaden, inte de enskilda spinntillstånden. Att skapa en sådan algoritm krävde att de släppte antaganden som utvecklats från många års användning av konventionella datorer och fokuserade på de unika egenskaperna hos kvantberäkning - nämligen "kvantöverlagringstillstånd".
"Superposition" låter algoritmer representera två variabler samtidigt, som sedan tillåter forskare att fokusera på förhållandet mellan dessa variabler utan att behöva bestämma sina individuella tillstånd först. Forskargruppen använde något som kallas en bruten symmetri-vågfunktion som en superposition av vågfunktioner med olika spinntillstånd och skrev om den till den Hamiltonianska ekvationen för parametern J. Genom att köra denna nya kvantkrets, laget kunde fokusera på avvikelser från sitt mål och genom att tillämpa Bayesian slutsats, en maskininlärningsteknik, de tog in dessa avvikelser för att bestämma utbytesinteraktionsparametern J. "Numeriska simuleringar baserade på denna metod utfördes för kovalent dissociation av molekylärt väte (H 2 ), trippelbindningsdissociationen av molekylärt kväve (N 2 ), och marktillstånden för C, O, Si -atomer och NH, ÅH + , CH 2 , NF och O 2 molekyler med ett fel på mindre än 1 kcal/mol, "tillägger forskargruppen.
"Vi planerar att installera vår Bayesian eXchange-kopplingsparameterkalkylator med programvara för Broken-symmetri-vågfunktioner (BxB) på korttidskvantdatorer utrustade med bullriga (ingen kvantfelkorrigering) mellanskala (flera hundra qubits) kvantanordningar (NISQ-enheter ), testa användbarheten för kvantkemiska beräkningar av faktiska stora molekylsystem. "