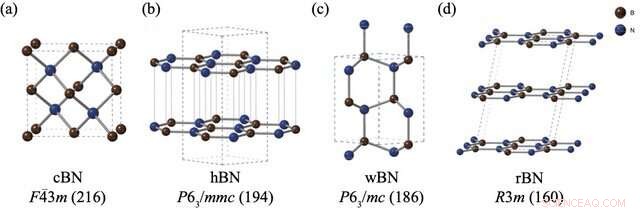
Strukturerna och rymdgrupperna av (a) zinkblandning bornitrid (cBN), (b) hexagonal bornitrid (hBN), (c) wurtzite bornitrid (wBN) och (d) romboedrisk bornitrid (rBN). Bor- och kväveatomer är avbildade i brunt respektive blått. Kredit:Kousuke Nakano från JAIST.
Bornitrid (BN) är ett mångsidigt material med tillämpningar inom en mängd olika tekniska och vetenskapliga områden. Detta beror till stor del på en intressant egenskap hos BN som kallas "polymorfism", kännetecknad av förmågan att kristallisera till mer än en typ av struktur. Detta sker vanligtvis som ett svar på förändringar i temperatur, tryck eller båda. Dessutom skiljer sig de olika strukturerna, kallade "polymorfer", anmärkningsvärt i sina fysikaliska egenskaper trots att de har samma kemiska formel. Som ett resultat spelar polymorfer en viktig roll i materialdesign, och kunskap om hur man selektivt gynnar bildningen av den önskade polymorfen är avgörande i detta avseende.
Emellertid utgör BN-polymorfer ett särskilt problem. Trots att flera experiment genomförts för att bedöma den relativa stabiliteten hos BN-polymorfer, har det inte uppstått någon konsensus om detta ämne. Medan beräkningsmetoder ofta är det vanligaste tillvägagångssättet för dessa problem, har BN-polymorfer ställt till allvarliga utmaningar för standardberäkningstekniker på grund av de svaga "van der Waals (vdW) interaktionerna" mellan deras lager, vilket inte tas med i dessa beräkningar. Dessutom manifesterar de fyra stabila BN-polymorferna, nämligen romboedriska (rBN), hexagonala (hBN), wurtzite (wBN) och zink-blandning (cBN), inom ett smalt energiområde, vilket gör att små energiskillnader fångas upp tillsammans med vdW-interaktioner ännu mer utmanande.
En internationell forskargrupp ledd av biträdande professor Kousuke Nakano från Japan Advanced Institute of Science and Technology (JAIST) har nu tillhandahållit bevis för att lösa debatten. I sin studie tog de upp problemet med ett toppmodernt ramverk för beräkning av första principer, nämligen Monte Carlo-simuleringar med fix nod (FNDMC). FNDMC representerar ett steg i den populära kvantmetoden Monte Carlo, där en parametriserad kvantvågfunktion för många kroppar först optimeras för att uppnå grundtillståndet och sedan tillförs FNDMC.
Dessutom beräknade teamet också Gibbs-energin (det användbara arbete som kan erhållas från ett system vid konstant tryck och temperatur) för BN-polymorfer för olika temperaturer och tryck med hjälp av densitetsfunktionsteori (DFT) och fononberäkningar. Denna artikel gjordes tillgänglig online den 24 mars 2022 publicerad i The Journal of Physical Chemistry C .
Enligt FNDMC-resultaten var hBN den mest stabila strukturen, följt av rBN, cBN och wBN. Dessa resultat var konsekventa vid både 0 K och 300 K (rumstemperatur). DFT-uppskattningarna gav dock motstridiga resultat för två olika uppskattningar. Dr. Nakano förklarar dessa motsägelsefulla fynd:"Våra resultat avslöjar att uppskattningen av relativa stabiliteter i hög grad påverkas av utbytets korrelationell funktion, eller den approximation som används i DFT-beräkningen. Som ett resultat kan en kvantitativ slutsats inte nås med DFT-fynd, och ett mer exakt tillvägagångssätt, såsom FNDMC, krävs."
Noterbart var FNDMC-resultaten i överensstämmelse med de som genererades av andra raffinerade beräkningsmetoder, såsom "kopplat kluster", vilket tyder på att FNDMC är ett effektivt verktyg för att hantera polymorfer, särskilt de som styrs av vdW-krafter. Teamet visade också att det kan tillhandahålla annan viktig information, såsom tillförlitliga referensenergier, när experimentella data inte är tillgängliga.
Dr. Nakano är exalterad över metodens framtidsutsikter inom materialvetenskap. "Vår studie visar FNDMCs förmåga att upptäcka små energiförändringar som involverar vdW-krafter, vilket kommer att stimulera användningen av denna metod för andra van der Waals-material", säger han. "Dessutom kan molekylära simuleringar baserade på denna exakta och tillförlitliga metod möjliggöra materialdesign, vilket möjliggör utveckling av läkemedel och katalysatorer." + Utforska vidare