Nya aktiva läkemedelsingredienser lägger grunden för innovativa och bättre medicinska behandlingar. Att identifiera dem och framförallt producera dem genom kemisk syntes i laboratoriet är dock ingen enkel bedrift. För att komma in på den optimala produktionsprocessen använder kemister vanligtvis en trial-and-error-metod:de härleder möjliga metoder för laboratoriesyntes från kända kemiska reaktioner och testar sedan var och en med experiment, ett tidskrävande tillvägagångssätt som är full av återvändsgränder .
Nu har forskare vid ETH Zürich, tillsammans med forskare från Roche Pharma Research and Early Development, kommit fram till ett tillvägagångssätt baserat på artificiell intelligens som hjälper till att bestämma den bästa syntesmetoden, inklusive dess sannolikhet för framgång. Deras artikel är publicerad i tidskriften Nature Chemistry .
"Vår metod kan avsevärt minska antalet laboratorieexperiment som krävs", förklarar Kenneth Atz, som utvecklade AI-modellen som doktorand tillsammans med professor Gisbert Schneider vid Institute of Pharmaceutical Sciences vid ETH Zürich.
Aktiva farmaceutiska ingredienser består vanligtvis av en ställning på vilken är bundna så kallade funktionella grupper. Det är dessa som ger ämnet dess mycket specifika biologiska funktion. Ställningens uppgift är att föra de funktionella grupperna i en definierad geometrisk linjering så att de kan agera målinriktat. Föreställ dig en kranbyggsats, i vilken ett ramverk av kopplingselement är bultade ihop på ett sådant sätt att funktionella enheter som rullar, kabelvinschar, hjul och förarhytten är korrekt anordnade i förhållande till varandra.
Ett sätt att producera läkemedel med en ny eller förbättrad medicinsk effekt är att placera funktionella grupper på nya platser på ställningarna. Detta kan låta enkelt, och det skulle verkligen inte utgöra ett problem på en modellkran, men det är särskilt svårt i kemi. Detta beror på att byggnadsställningarna, som huvudsakligen består av kol- och väteatomer, i sig är praktiskt taget icke-reaktiva, vilket gör det svårt att binda dem med funktionella atomer som syre, kväve eller klor. För att detta ska lyckas måste ställningarna först aktiveras kemiskt via omvägsreaktioner.
En aktiveringsmetod som öppnar väldigt många möjligheter för olika funktionella grupper, åtminstone på pappret, är borylering. I denna process är en kemisk grupp som innehåller grundämnet bor bunden till en kolatom i ställningen. Borgruppen kan då helt enkelt ersättas av en hel rad medicinskt effektiva grupper.
"Även om borylering har stor potential, är reaktionen svår att kontrollera i labbet. Det är därför som vår omfattande sökning av den världsomspännande litteraturen endast visade drygt 1 700 vetenskapliga artiklar om ämnet", säger Atz och beskriver utgångspunkten för sitt arbete.
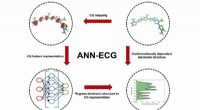
Tanken var att ta reaktionerna som beskrivs i den vetenskapliga litteraturen och använda dem för att träna en AI-modell, som forskargruppen sedan kunde använda för att överväga nya molekyler och identifiera så många platser som möjligt på dem där borylering skulle vara möjligt. Men forskarna matade i slutändan sin modell endast med en bråkdel av den litteratur de hittade. För att säkerställa att modellen inte vilseleds av falska resultat från slarvig forskning, begränsade teamet sig till 38 särskilt pålitliga artiklar. Dessa beskrev totalt 1 380 boryleringsreaktioner.
För att utöka träningsdatauppsättningen kompletterade teamet litteraturresultaten med utvärderingar av 1 000 reaktioner utförda i det automatiserade laboratoriet som drivs av Roches forskningsavdelning för medicinsk kemi. Detta gör att många kemiska reaktioner kan utföras på milligramskalan och analyseras samtidigt.
"Att kombinera laboratorieautomation med AI har en enorm potential för att kraftigt öka effektiviteten i kemisk syntes och förbättra hållbarheten på samma gång", säger David Nippa, doktorand från Roche som genomförde projektet tillsammans med Atz.
De prediktiva kapaciteterna hos modellen som genererades från denna datapool verifierades med hjälp av sex kända läkemedelsmolekyler. I 5 av 6 fall bekräftade experimentell testning i laboratoriet de förutspådda ytterligare platserna. Modellen var lika pålitlig när det gällde att identifiera platser på ställningen där aktivering inte är möjlig. Dessutom bestämde den de optimala förhållandena för aktiveringsreaktionerna.
Intressant nog blev förutsägelserna ännu bättre när 3D-information om utgångsmaterialen inkluderades snarare än bara deras tvådimensionella kemiska formler. "Det verkar som om modellen utvecklar en sorts tredimensionell kemisk förståelse", säger Atz.
Framgångsfrekvensen för förutsägelserna imponerade också på forskarna vid Roche Pharma Research and Early Development. Under tiden har de framgångsrikt använt metoden för att identifiera platser i befintliga läkemedel där ytterligare aktiva grupper kan introduceras. Detta hjälper dem att snabbare utveckla nya och mer effektiva varianter av kända aktiva läkemedelsingredienser.
Atz och Schneider ser många andra möjliga tillämpningar för AI-modeller som är baserade på en kombination av data från pålitlig litteratur och från experiment utförda i ett automatiserat laboratorium. Detta tillvägagångssätt borde till exempel göra det möjligt att skapa effektiva modeller för andra aktiveringsreaktioner än borylering. Teamet hoppas också kunna identifiera ett bredare spektrum av reaktioner för att ytterligare funktionalisera de borylerade platserna.
Atz är nu involverad i detta vidareutvecklingsarbete som AI-forskare inom läkemedelskemi vid Roche. "Det är väldigt spännande att arbeta i gränssnittet mellan akademisk AI-forskning och laboratorieautomation. Och det är ett nöje att kunna driva detta framåt med det bästa innehållet och metoderna", säger Atz.
Schneider tillägger, "Detta innovativa projekt är ytterligare ett enastående exempel på samarbete mellan akademi och industri och visar den enorma potentialen hos offentlig-privata partnerskap för Schweiz."
Mer information: David F. Nippa et al, möjliggöra läkemedelsdiversifiering i sent skede genom experiment med hög genomströmning med geometrisk djupinlärning, Nature Chemistry (2023). DOI:10.1038/s41557-023-01360-5
Journalinformation: Naturkemi
Tillhandahålls av ETH Zürich