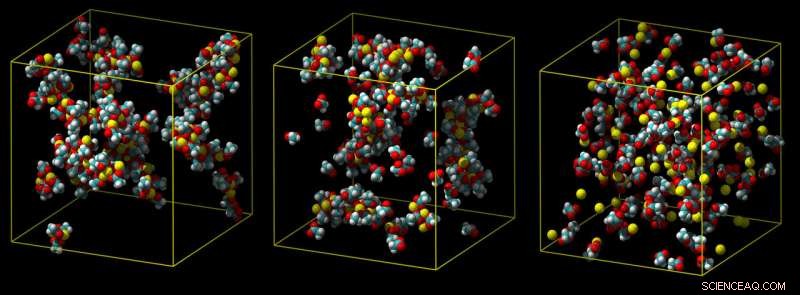
En standard kalciummodell överskattar hur starkt kalcium binder, vilket leder till klumpar av jonpar (vänster). En mellanliggande modell visar mindre klumpning (mitten), och en förfinad laddningsskalemodell förutsäger korrekt en svag koppling till karboxylgrupper i vatten (visas inte) (höger). Upphovsman:Philip Mason och Elise Duboue-Dijon
Kalcium är viktigt för att våra kroppar ska fungera. Kalciumjoner gör att celler kan kommunicera med varandra, låter neuroner interagera, muskler att dra ihop sig, och hjärtats muskelceller för att synkronisera och slå. För att bättre förstå dessa processer, i vilka kalciumjoner interagerar med biologiska molekyler som proteiner, forskare använder ofta datasimuleringar. Men exakta modeller är utmanande och beräkningsmässigt dyra.
"Om du har fel modell av kalcium, det kommer helt enkelt inte att fungera, "sade Pavel Jungwirth vid Institutet för organisk kemi och biokemi vid Tjeckiska vetenskapsakademien i Prag." De flesta modeller som finns tillgängliga är inte tillräckligt noggranna för att fånga de viktiga egenskaperna hos kalciumjonen. "
I veckans nummer av Journal of Chemical Physics , dock, Jungwirths forskargrupp visar hur en enkel modifiering i en datormodell leder till mycket exakta simuleringar, som fungerar som kraftfulla verktyg för att studera en rad biologiska processer. "Jag tror att vi har de bästa av de enkla kalciummodellerna i världen för tillfället, "Sa Jungwirth.
Kalciumjoner reser från cell till cell som budbärare. När de når en cell, de binder till en molekyl, som ett protein, utlöser en kaskad av kemiska svar. Men på grund av jonens vattniga miljö, det är svårt att simulera exakt hur kalcium binder.
Kalciumjonen, som är dubbelt positivt laddad, interagerar starkt med oxigenerna i de omgivande vattenmolekylerna. Dessa oxygens har en partiell negativ laddning (som i vattenmolekylen) och syreatomen lockar elektronerna i bindningarna mer effektivt. De elektrostatiska krafterna mellan kalcium och vatten får vattenmolekylerna att omarrangera sig runt jonen. Kalciumjonen tvingar också elektronerna i vattenmolekylen att skifta, ett fenomen som kallas elektronisk polarisering.
De flesta simuleringar innehåller omläggning av vattenmolekyler. Men för att beräkna exakt hur elektroner rör sig kräver för mycket datorkraft, de tar inte hänsyn till elektronisk polarisering. Utan elektronisk polarisering, Jungwirth sa, simuleringar med kalcium är felaktiga.
Vanligtvis, interaktioner med vattenmolekyler arbetar för att dra en kalciumjon bort från molekylen den försöker binda med, som i en molekylär dragkamp. Om en simulering inte helt tar hänsyn till dessa effekter, det överskattar hur starkt kalcium binder, producerar joner som inte kan avbindas, vilket är orealistiskt.
Några år sedan, dock, Alexei Stuchebrukhov och Igor Leontyev föreslog en lösning:Sänk jonernas elektriska laddning i simuleringarna. Det visar sig att skalning av laddningen med en faktor på cirka 0,75 efterliknar effekten av elektronisk polarisering. En sådan enkel skalning lägger inte heller till någon extra beräkningsbörda.
"Det är nästan ett mirakel, "Sa Jungwirth." Vi vet att det inte är en perfekt lösning, men kanske löser det 90 procent av problemet. "
Tidigare, Jungwirths team testade strategin genom att modellera den relativt enkla interaktionen mellan kalcium- och kloridjoner. För att kontrollera om simuleringarna var korrekta - och om skalningen fungerade - sprängde de riktiga kalciumkloridlösningar med neutroner. Genom att mäta hur dessa neutroner sprids från den vattenhaltiga kalciumkloriden, forskarna härledde dess struktur och jämförde data med simuleringarna.
I den nya studien, forskarna testade sin modell med karboxylgrupper - molekylära grupper som finns i proteiner, och därmed mer relevant för biologin. Efter att även ha justerat laddningen för karboxylgruppen, de visade återigen att deras simuleringar matchade mycket bra med data från neutronspridningsexperiment.
Eftersom karboxylgrupper är enkla jämfört med, säga, ett helt protein, forskarna kunde också beskriva kalciuminteraktionerna med hjälp av exakta men beräkneligt dyra elektroniska strukturberäkningar. Genom att jämföra dessa beräkningar med simuleringarna, de bekräftade återigen riktigheten i sina modeller.
Dessa tester visar att den nya modellen kan simulera kalciuminteraktioner med nästan vilket protein som helst, Sa Jungwirth. Forskarna har också utvecklat en analog modell som fungerar för kalciuminteraktioner med fosfolipider vid cellmembranet. Nästa steg, han sa, är att göra samma sak med DNA- och RNA -molekyler. Och längre fram, forskarna planerar att utveckla en liknande modell för magnesium, en annan viktig signaljon med sina egna unika utmaningar.