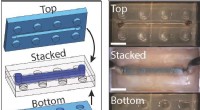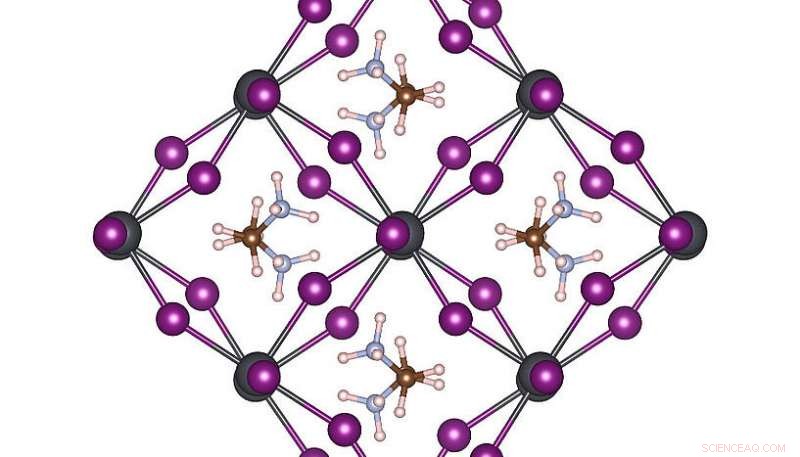
Hög symmetri atomstruktur av MAPbI3 vid rumstemperatur. Upphovsman:Menno Bokdam/Universitetet i Wien
På atomskala kan material visa en rik palett av dynamiskt beteende, som direkt påverkar de fysikaliska egenskaperna hos dessa material. Under många år, det har varit en dröm att beskriva denna dynamik i komplexa material vid olika temperaturer med hjälp av datasimuleringar. Fysiker vid universitetet i Wien har utvecklat en in-the-fly maskininlärningsmetod som möjliggör sådana beräkningar genom direkt integration i den kvantmekanikbaserade Vienna Ab-initio Simulation Package (VASP). Självlärningsmetodens mångsidighet demonstreras av nya fynd, publicerad i tidningen Fysiska granskningsbrev , på fasövergångarna av hybridperovskiter. Dessa perovskiter är av stort vetenskapligt intresse på grund av deras potential inom skörd av solenergi och andra tillämpningar.
Vid rumstemperatur, alla material rör sig ständigt i atomskala. Även fast sten består av atomer som svänger runt. Materialens fysikaliska egenskaper är direkt kopplade till arrangemanget av atomer i, så kallade, kristallgitter. Beroende på temperaturen eller trycket kan detta arrangemang förändras och därmed påverka materialegenskaperna. Man kan tänka på diamant, som är transparent och hårt på grund av det periodiska arrangemanget av kolatomer i diamantkristallen. Samma atomer, ordnade annorlunda, resulterar i svart, spröd grafit. Det var redan möjligt att exakt beräkna koordinaterna för atomerna i enkla material vid olika temperaturer med kvantmekaniska molekylära dynamiska (MD) simuleringar. Dock, sådana beräkningar är beräknat dyra och begränsar praktiska tillämpningar till ett par hundratals atomer och begränsad simuleringstid.
Fysiker från Computational Materials Physics -gruppen vid universitetet i Wien har utvecklat ett nytt tillvägagångssätt som övervinner dessa begränsningar och möjliggör simuleringar av komplexa material för framtida energitillämpningar. Detta uppnås genom att utveckla en effektiv och robust datadriven självlärande algoritm och, viktigast, genom att integrera denna algoritm direkt i Wien Ab-initio Simulation Package (VASP). I det nya tillvägagångssättet, "maskinen" kan ta upp, på egen hand, de viktigaste ingredienserna för en enklare modellbeskrivning av de interagerande atomerna under MD -simuleringar. Redan efter att ha beräknat några hundratals tidssteg kan maskinen förutsäga tillräckligt noga atomernas positioner i det på varandra följande tidssteget. Maskinen kan också göra en uppskattning av dess noggrannhet för de på varandra följande stegen. Om felet är för högt, maskinen växlar och utför det mycket exakta, men dyrt, MD -beräkningar. Ju mer simuleringstid som går ju mer maskinen lär sig och desto mer exakt blir den. På det här sättet, färre och färre MD -beräkningar krävs, vilket så småningom leder till situationen där alla tidsteg görs av maskinen. Dessutom, den on-the-fly självlärande förmågan minskar behovet av mänskligt ingripande som krävs av andra befintliga maskininlärningsmetoder.
För att visa kraften i denna nya metod, forskarna har tillämpat den för att studera övergångarna mellan olika atomstrukturer i MAPbI 3 perovskit vid ändring av temperaturen. Detta material är mycket populärt på grund av dess potential som en ny billig solcellskomponent. Den är gjord av organiska molekyler som snabbt kan vända, separerade från varandra med ett gitter bestående av bly- och jodidatomer. Beroende på temperaturen bildas tre olika kristallfaser. Atommekanismerna nära övergångstemperaturen är mycket svåra att bestämma genom experiment, och MD-simuleringar skulle kräva år med beräkningstid även på ett modernt superdatasystem. Efter att ha lärt sig, maskinen kan förutsäga fasövergångstemperaturer och gitterkonstanter av detta material med en aldrig tidigare skådad precision. Den utvecklade metoden är allmän och tillämplig på många andra framtida materialvetenskapliga problem och kommer att bli tillgänglig för forskare ordomfattande i den kommande versionen av VASP.
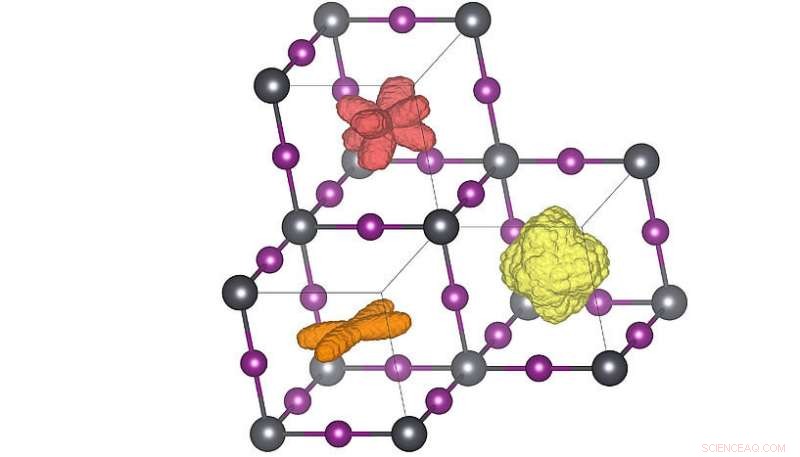
Tredimensionella fördelningar av molekylens orientering i de tre olika kristallfaserna. När temperaturen höjs (orange → röd → gul) kan molekylerna uppnå fler riktningar. Den röda fördelningen motsvarar rumstemperaturstrukturen. Upphovsman:Menno Bokdam/Universitetet i Wien