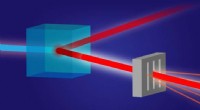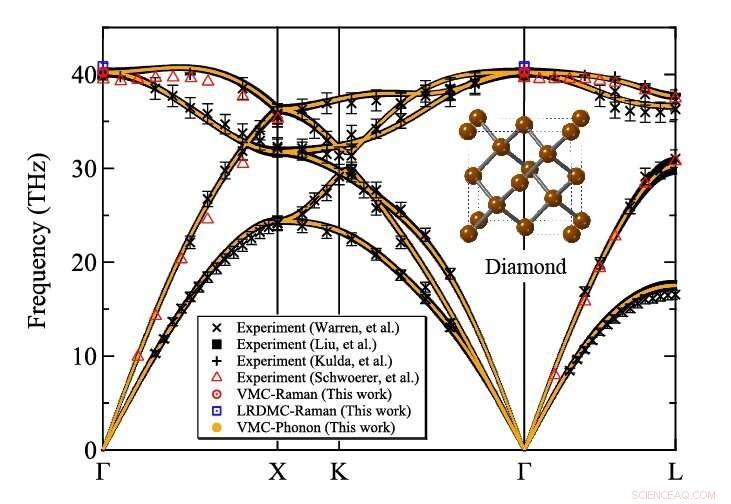
Phonon -spridning av diamant beräknat på Monte Carlo -variationen av TurboRVB. Upphovsman:Kousuke Nakano från JAIST
Fokus och slutmål för beräkningsforskning inom materialvetenskap och kondenserad materiens fysik är att lösa Schrödinger -ekvationen - den grundläggande ekvationen som beskriver hur elektroner beter sig inuti materia - exakt (utan att behöva förenkla approximationer). Även om experiment verkligen kan ge intressanta inblickar i materialets egenskaper, det är ofta beräkningar som avslöjar den bakomliggande fysiska mekanismen. Dock, beräkningar behöver inte förlita sig på experimentella data och kan, faktiskt, utföras självständigt, en metod som kallas "ab initio -beräkningar". Densitetsfunktionell teori (DFT) är ett populärt exempel på ett sådant tillvägagångssätt.
För de flesta materialvetare och fysiker av kondenserade ämnen, DFT -beräkningar är brödet och smöret i deras yrke. Dock, trots att det är en kraftfull teknik, DFT har haft begränsad framgång med "starkt korrelerade material" - material med ovanliga elektroniska och magnetiska egenskaper. Dessa material, medan de är intressanta på egen hand, har också tekniska användbara egenskaper, ett faktum som starkt motiverar ett ab initio -ramverk som är lämpligt att beskriva dem.
För detta ändamål, en ram som kallas "ab initio quantum Monte Carlo" (QMC) har visat betydande lovande och förväntas bli nästa generation av elektroniska strukturberäkningar på grund av dess överlägsenhet gentemot DFT. Dock, även QMC är i stort sett begränsat till beräkningar av energi och atomkrafter, begränsa dess användbarhet vid beräkning av användbara materialegenskaper.
Nu, i en genombrottstudie publicerad i Fysisk granskning B (Redaktörernas förslag), forskare har tagit saker till nästa nivå baserat på ett tillvägagångssätt som gör att de kan minska det statistiska felet vid atomkraftsutvärdering med två storleksordningar och därefter påskyndar beräkningen med en faktor 10 4 ! "Den drastiska minskningen av beräkningstiden kommer att kraftigt utöka intervallet för QMC -beräkningar och möjliggöra mycket exakt förutsägelse av atomegenskaper för material som har varit svåra att hantera, "konstaterar biträdande professor Kousuke Nakano från Japan Advanced Institute of Science and Technology (JAIST), WHO, tillsammans med sina kollegor prof. Ryo Maezono från JAIST, Prof. Sandro Sorella från International School for Advanced Studies (SISSA), Italien, och Dr. Tommaso Morresi och prof. Michele Casula från Sorbonne Université, Frankrike, ledde denna banbrytande prestation.
Teamet tillämpade sin utvecklade metod för att beräkna diamantens atomvibrationer, ett typiskt referensmaterial, som ett bevis på konceptet och visade att resultaten överensstämde med experimentella värden. För att utföra dessa beräkningar, de använde en stor dator, Cray-XC40, ligger vid Research Center for Advanced Computing Infrastructure vid Japan Advanced Institute of Science and Technology (JAIST), Japan, tillsammans med en annan på RIKEN, Japan. Teamet använde ett QMC -mjukvarupaket som heter "TurboRVB, "ursprungligen lanserades av prof. Sorella och prof. Casula och utvecklades senare av prof. Nakano tillsammans med andra, att utföra beräkningar av fonondispersion för diamant som tidigare var otillgängliga, utvidgar omfattningen kraftigt.
Prof. Nakano ser fram emot tillämpningarna av QMC inom materialinformatik (MI), ett fält dedikerat till design och sökning efter nya material med hjälp av tekniker för informationsvetenskap och beräkningsfysik. "Medan MI för närvarande styrs av DFT, den snabba utvecklingen av datorprestanda, till exempel exascale superdator, hjälper QMC att bli populär. I det avseendet, vår utvecklade metod kommer att vara mycket användbar för att designa nya material med verkliga applikationer, "avslutar en optimistisk doktor Nakano.