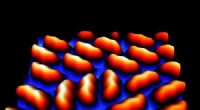
Nanoporen begränsar rörelsefriheten för den adsorberade enstaka molekylen, vilket gör det möjligt för forskare vid Technische Universitat Munchen och University Lingkoping att modellera enstaka molekylers jämviktstermodynamik. Upphovsman:Carlos-Andres Palma / TUM
Högpresterande material för gaslagring, värmeisolatorer eller nanomaskiner behöver en grundlig förståelse av materialets beteende ner till molekylär nivå. Termodynamik, som har utvecklats för två hundra år sedan för att öka effektiviteten hos ångmotorer, observerar och medelvärderar vanligtvis över ett stort antal molekyler. Nu har ett team av forskare utvecklat en metodik, att undersöka jämviktstermodynamiken för enstaka molekyler.
På jakt efter högpresterande material för applikationer som gaslagring, värmeisolatorer eller dynamiska nanosystem är det viktigt att förstå materiens termiska beteende ner till molekylär nivå. Klassisk termodynamik genomsnitt över tid och över ett stort antal molekyler. Inom ett tredimensionellt utrymme kan enstaka molekyler anta ett nästan oändligt antal tillstånd, gör bedömningen av enskilda arter nästan omöjlig.
Nu har forskare från Technische Universität München (TUM) och Linköpings universitet (LIU) utvecklat en metodik, vilket gör det möjligt att utforska jämviktstermodynamik för enstaka molekyler med atomupplösning vid märkbara temperaturer. Genombrottsstudien bygger på två pelare:en teknik som gör det möjligt att hålla kvar molekyler inom tvådimensionella nanoporer och omfattande beräkningsmodeller.
Fångad i två dimensioner
Vid ordförande för molekylär nanovetenskap och kemisk fysik i gränssnitt vid TU München, ledd av prof. dr. Johannes V. Barth, PD Dr Florian Klappenberger utvecklade metoden för att producera högkvalitativa metall-organiska nätverk på en silveryta. Nätverket bildar nanoporer som begränsar rörelsefriheten för adsorberade enstaka molekyler i två dimensioner. Med hjälp av skanningstunnelmikroskopi kunde forskarna spåra sina rörelser vid olika temperaturer med sub-nanometerupplösning.
Parallellt med experimenten, forskarna arbetade med sofistikerade datormodeller för att beskriva temperaturberoendet för dynamiken i dessa enstaka fångade molekyler. "Vi har tillämpat toppmoderna superdatorberäkningar för att förstå interaktioner och energilandskap som bestämmer molekylernas rörelse", säger Jonas Björk från Linköpings universitet.
Genom att jämföra experimentella och modellerade data upptäckte forskarna att den integrerade teorin under vissa förhållanden närmar sig en enkel projicering av de molekylära positionerna i rymden. Detta tillvägagångssätt är centralt för statistisk mekanik, men har aldrig tidigare utmanats att återge ett experiment, på grund av de praktiskt taget oändliga molekylära positionerna och energierna som man behövde tänka på utan nanoskala.
Analogi till biologi
"Det var oerhört spännande att använda tvådimensionella nätverk som en inneslutningsstrategi för att minska tillgängligt konformationsutrymme för en enda molekyl, som en chaperone gör med ett protein ", säger Dr Carlos-Andres Palma, huvudförfattaren till studien. "I analogi med biologi, sådan form av inneslutningsteknik har potential att etablera sensorer, nanomaskiner och möjligen logiker som styrs av och består av molekylära fördelningar. "
Tillämpa sin kunskap om karakteristiska jämviktskonfigurationer, forskarna modulerade noggrant nanoporen, vilket får en enda molekyl att skriva bokstäver i alfabetet som L, Jag och U, bara genom att finjustera temperaturen.