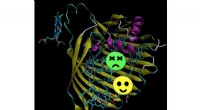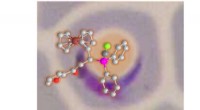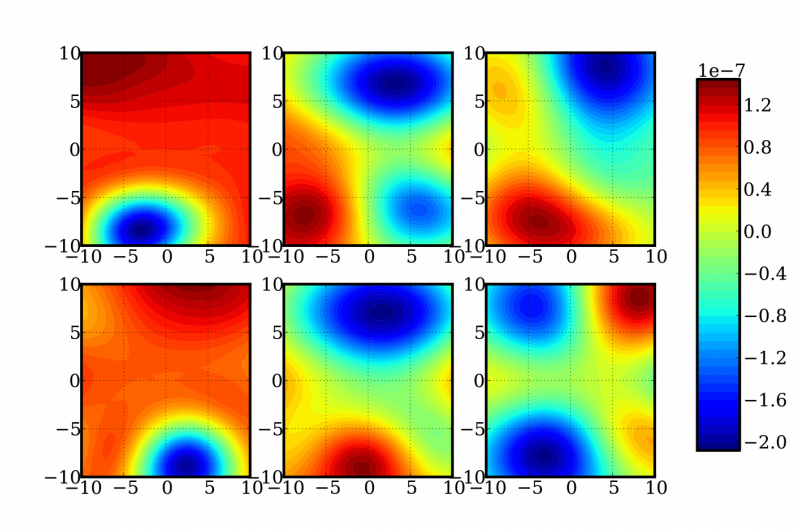
En skildring av slumpmässiga tvådimensionella skivor av en 12-dimensionell funktion för bestämning av energi- och frekvenskorrigeringar av en formaldehydmolekyl. Upphovsman:Sandia National Laboratories
Forskare vid Sandia National Laboratories har utvecklat nya matematiska tekniker för att främja studiet av molekyler på kvantnivå.
Matematisk och algoritmisk utveckling längs dessa linjer är nödvändiga för att möjliggöra detaljerad studie av komplexa kolvätemolekyler som är relevanta vid motorförbränning.
Befintliga metoder för att approximera potentiella energifunktioner i kvantskalan behöver för mycket datorkraft och är därför begränsade till små molekyler. Sandia -forskare säger att deras teknik kommer att påskynda kvantmekaniska beräkningar och förbättra förutsägelser från teoretiska kemimodeller. Med tanke på beräkningshastigheten, dessa metoder kan eventuellt tillämpas på större molekyler.
Sandia postdoktorala forskare Prashant Rai arbetade med forskarna Khachik Sargsyan och Habib Najm vid Sandias förbränningsforskningsanläggning och samarbetade med kvantkemister So Hirata och Matthew Hermes vid University of Illinois i Urbana-Champaign. Beräkningsenergi vid färre geometriska arrangemang än normalt krävs, laget utvecklade beräkningseffektiva metoder för att approximera potentiella energiytor.
En exakt förståelse av potentiella energiytor, nyckelelement i praktiskt taget alla beräkningar av kvantdynamik, krävs för att exakt uppskatta energin och frekvensen för molekylernas vibrationssätt.
"Om vi kan hitta molekylens energi för alla möjliga konfigurationer, vi kan bestämma viktig information, såsom stabila tillstånd för molekylär övergångsstruktur eller mellanliggande tillstånd för molekyler i kemiska reaktioner, "Sa Rai.
De första resultaten av denna forskning publicerades i Molekylär fysik i en artikel med titeln "Low-rank canonical-tensor sönderdelning av potentiella energiytor:applikation till nätbaserad diagrammatisk vibration Greens funktionsteori."

Sandia National Laboratories forskare Prashant Rai, vänster, Habib Najm, Centrum, och Khachik Sargsyan diskuterar matematiska tekniker som används för att studera beteendet hos stora molekyler i kvantskala. Upphovsman:Dino Vournas
"Att approximera potentiella energiytor hos större molekyler är en extremt utmanande uppgift på grund av den exponentiella ökningen av information som krävs för att beskriva dem med varje ytterligare atom i systemet, "Sa Rai." I matematik, det kallas dimensionens förbannelse. "
Slog förbannelsen
Nyckeln till att slå förbannelsen över dimensionalitet är att utnyttja egenskaperna hos den specifika strukturen hos de potentiella energiytorna. Rai sa att denna strukturinformation sedan kan användas för att approximera de nödvändiga högdimensionella funktionerna.
"Vi använder det faktum att även om potentiella energiytor kan vara högdimensionella, de kan väl approximeras som en liten summa produkter med endimensionella funktioner. Detta är känt som den låga strukturen, där den potentiella energiytans rang är antalet termer i summan, "Rai sa." Ett sådant antagande om struktur är ganska allmänt och har också använts i liknande problem på andra områden. Matematiskt, intuitionen av lågt rankade approximationstekniker kommer från flerclinig algebra där funktionen tolkas som en tensor och sönderdelas med hjälp av standard tensors sönderdelningstekniker. "
Energi- och frekvenskorrigeringarna formuleras som integraler av dessa högdimensionella energifunktioner. Tillnärmning i ett sådant lågt rankat format gör dessa funktioner lätt integrerbara eftersom det bryter integrationsproblemet till summan av produkter från en- eller tvådimensionella integraler, så vanliga integrationsmetoder gäller.
Teamet testade sina beräkningsmetoder på små molekyler som vatten och formaldehyd. Jämfört med den klassiska Monte Carlo -metoden, slumpmässigt baserad standard arbetshäst för högdimensionella integrationsproblem, deras tillvägagångssätt förutsade energi och frekvens för vattenmolekyl som var mer exakta, och det var minst 1, 000 gånger mer beräkningseffektivt.
Rai sa att nästa steg är att ytterligare förbättra tekniken genom att utmana den med större molekyler, såsom bensen.
"Tvärvetenskapliga studier, såsom kvantkemi och förbränningsteknik, ge möjligheter till korsbestämning av idéer, därigenom ge ett nytt perspektiv på problem och deras möjliga lösningar, "Rai sa." Det är också ett steg mot att använda de senaste framstegen inom datavetenskap som en pelare för vetenskaplig upptäckt i framtiden. "