Ammoniak (NH3 ) är en viktig molekyl med många tillämpningar. Slutprodukten av den berömda Haber-Bosch-processen, den syntetiseras vanligtvis för att fånga upp kväve för gödningsmedel, och används för kylning, i rengöringsprodukter och vid tillverkning av läkemedel. På senare tid har denna blygsamma molekyl också väckt intresse som en potentiell resurs för att ta itu med en av dagens mest angelägna utmaningar – behovet av pålitliga och rikliga förnybara bränslen.
Ammoniak är stabil och säker att hantera, är brännbar och innehåller den största fraktionen väte av någon molekyl utom rent väte i sig. Dessa faktorer lovar att göra det till ett genomförbart alternativ till de kolbaserade energibärare som driver klimatförändringarna. Forskning har börjat undersöka hur ammoniak kan användas för att direkt driva motorer, gasturbiner och vätebränsleceller, till exempel. Man tror också att ammoniak skulle kunna användas för att lagra energi för tider då andra förnybara energikällor som vind- och solenergi inte kan möta efterfrågan.
Mycket är känt om ammoniak, men intresset för att använda det som bränsle har initierat ett sökande efter nya ammoniakteknologier. Detta har i sin tur lett till ett ökat behov bland kemiingenjörer av korrekta data som beskriver ammoniakens grundläggande termodynamiska egenskaper. Sådana egenskaper inkluderar en mängd olika mätbara egenskaper såsom fasjämvikter, densitet eller värmekapacitet, till exempel, som kännetecknar fysikaliska system och bestämmer hur kemiska processer fungerar. När det gäller ammoniak vill ingenjörer också ha bättre kunskap om hur sådana egenskaper förändras när man blandar ammoniak med andra molekyler. Sådan kunskap skulle kunna hjälpa dem att optimera processer och driftsförhållanden.
Dr. Jadran Vrabec, för närvarande chef för Institutet för processvetenskap vid Berlins tekniska universitet, har tillbringat mycket av sin karriär med att använda högpresterande beräkningar (HPC) för att undersöka termodynamiska egenskaper på molekylär nivå. "Termodynamiska egenskaper bestäms till 100% av molekylära interaktioner", förklarar han. "Och eftersom dessa interaktioner sker så snabbt och i så liten skala, är det bara möjligt att studera dem genom att utföra stora simuleringar med superdatorer."
I en nyligen publicerad artikel publicerad i Journal of Chemical &Engineering Data , han och medförfattaren Erich Mace vid TU Berlin rapporterar om resultaten av simuleringar fokuserade på de termodynamiska egenskaperna hos blandningar som innehåller ammoniak. Producerade med hjälp av superdatorn Hawk vid High-Performance Computing Center Stuttgart (HLRS), deras resultat lägger till värdefulla data som kan stödja utvecklingen av nya tillämpningar av ammoniak. Resultaten kan också hjälpa till att bedöma riktigheten av andra befintliga data, vilket säkerställer att ingenjörer har den bästa tillgängliga informationen för att arbeta med ämnet.
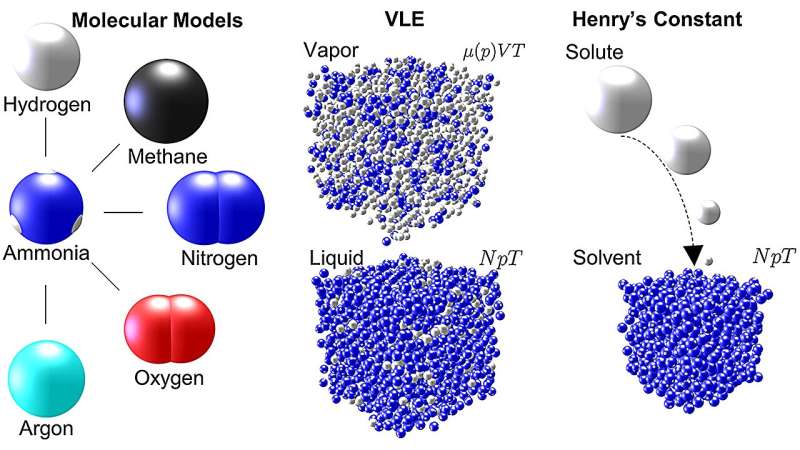
Storskaliga simuleringar ger unika insikter om termodynamiska egenskaper
Vrabec är en långvarig användare av HLRS superdatorresurser för molekylär dynamik och Monte Carlo-simuleringar. Hans tillvägagångssätt förlitar sig på termodynamikbegrepp som först artikulerades av Ludwig Boltzmann på 1800-talet, men som blev praktiska att tillämpa först på 1950-talet med ankomsten av de första datorerna. Sedan dess har området avancerat parallellt med utvecklingen av större och snabbare superdatorer, till den grad att Vrabecs simuleringar nu spårar individuella rörelser och interaktioner av miljarder eller till och med biljoner molekyler samtidigt. Med hjälp av programvara som hans labb utvecklat för att selektivt fånga in data av intresse kan han sedan studera molekylernas termodynamiska egenskaper.
Vrabec använder två simuleringskoder kallade ms2 och ls1, som han har utvecklat och optimerat under loppet av ett långt och fruktbart samarbete med HLRS-personalen Martin Bernreuther och Christoph Niethammer. Under 2019 satte teamet till och med ett världsrekord för det största molekylära systemet som någonsin simulerats med molekylära dynamikmetoder. Med hjälp av ls1 skalade de effektivt sin kod till ett system med 21 biljoner atomer där varje enskild molekyl och dess interaktioner med andra molekyler kunde spåras.
I det senaste arbetet med ammoniak utförde Mace och Vrabec molekylär dynamik och Monte Carlo-simuleringar med hjälp av ms2 för att undersöka fem vanliga blandningar som involverar ammoniak i kemiska processer:argon–ammoniak, metan–ammoniak, väte–ammoniak, kväve–ammoniak och syre -ammoniak. För varje blandning genererade simuleringarna data som beskriver ånga-vätska-jämvikten (VLE) – ett mått på fördelningen av molekyler i ett system över ång- eller vätskefaserna – för ett brett område av temperaturer och tryck.
I sin uppsats påpekar Mace och Vrabec att VLE-data ofta används för att utveckla tillståndsekvationer för industriella vätskor; det vill säga data kan användas för att förutsäga materiens tillstånd under olika fysiska förhållanden på grund av förändringar i temperatur, tryck, volym eller sammansättning. Sådan information är väsentlig för att bestämma optimala blandningar och arbetsförhållanden i industriella tillämpningar.
Vrabecs molekylära simuleringar är särskilt värdefulla eftersom de kan användas för att undersöka ett mycket bredare spektrum av skalor än vad som är möjligt med experimentella metoder.
"I våra simuleringar tillhandahöll vi mätningar av termodynamiska egenskaper även upp till tryck på 50 megapascal. Detta är 500 gånger vårt omgivande lufttryck," säger Vrabec. "Även om data för ammoniakblandningar har samlats in i mer än ett sekel är datatäckningen förvånansvärt snäv. Anledningen är att ansträngningen att mäta det experimentellt är oöverkomligt enorm. Det skulle kräva dyr specialutrustning som skulle vara farlig att använda. I datorsimuleringar kan vi få resultat säkert och relativt billigt." Hans metoder ger också en jämförbar nivå av noggrannhet med den för experimentella tillvägagångssätt inom områden där experimentella data finns tillgängliga.
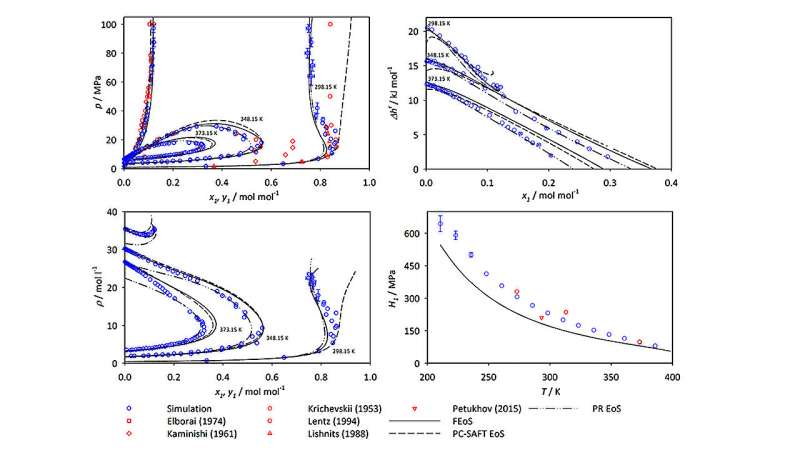
Bättre data för ammoniakforskning
När Mace och Vrabec analyserade sina simuleringsdata visade de att även om ammoniak är en komponent i alla fem system de studerade, ser de resulterande graferna av VLE-värden dramatiskt olika ut för olika molekylära blandningar. Enligt Vrabec, "Fasbeteendet hos olika blandningar bestäms starkt av interaktionerna mellan molekylerna i systemet. Du måste förstå dessa egenskaper om du är intresserad av att arbeta med ammoniakblandningar."
Tidningen och dess kompletterande data erbjuder mer än 400 nya datapunkter för varje blandning de studerade. Med hjälp av Hawk kunde de producera resultaten av varje blandning inom bara några dagars beräkningstid. Resultaten kommer att vara av särskilt värde för extrema, svårstuderade förhållanden för vilka lite data finns tillgänglig, och kan hjälpa ingenjörer att identifiera sweet spots där förhållandena skulle vara optimala för effektiv ammoniakbearbetning.
Studien inkluderade både nya simuleringsdata och tidigare publicerade data från den vetenskapliga litteraturen, vilket gjorde det möjligt för Mace och Vrabec att jämföra sina resultat med andra befintliga datauppsättningar av VLE-värden. I de flesta situationer överensstämde deras resultat nära med tidigare studier. I vissa fall identifierade de dock betydande skillnader mellan deras resultat och andra forskargruppers experimentellt genererade mätningar och förutsägelser. Författarna tillskriver dessa avvikelser till begränsningar eller felaktigheter i motsvarande experimentella metoder. De föreslår också att specifika experimentella datakällor bör användas med försiktighet i framtida forskning eller kemitekniska tillämpningar.
Vrabec säger att han i det senaste arbetet främst har fokuserat på att simulera termodynamiska egenskaper hos molekylära system, vanligtvis på submikrometerskala. Trots de många storleksordningar som ligger mellan denna skala och nivån av observerbara processer, finns det korrekta metoder för att översätta dessa insikter på molekylär nivå till användbara förutsägelser i den verkliga världen.
När superdatorer växer sig större, förutser han dock att det också kan bli möjligt att simulera inte bara egenskaper utan även termodynamiska processer med hjälp av randvillkor som ligger nära verkliga tillämpningar. Ökad HPC-prestanda kan ge mer exakta resultat om dynamiska fenomen med ett bättre signal-brusförhållande.
Under tiden visar hans teams resultat dock värdet av molekylär dynamik och Monte Carlo-simulering med hjälp av högpresterande beräkningar, och kommer att ge ny förståelse för fasbeteende som ingenjörer kan använda för att utveckla ny ammoniakbaserad teknik.
Mer information: Erich J. Mace et al, High-Pressure Fluid-Phase Equilibria and Henry's Constants of Supercritical Gases in Ammonia, Journal of Chemical &Engineering Data (2023). DOI:10.1021/acs.jced.3c00327
Tillhandahålls av Gauss Center for Supercomputing