En forskargrupp från Institutet för organisk kemi och biokemi vid Tjeckiska vetenskapsakademin / IOCB Prag har utvecklat en ny beräkningsmetod som exakt kan beskriva hur proteiner interagerar med molekyler av potentiella läkemedel och kan göra det på bara tiotals minuter. Denna nya kvantmekaniska poängfunktion kan således markant påskynda sökandet efter nya läkemedel. Forskningen har publicerats i tidskriften Nature Communications .
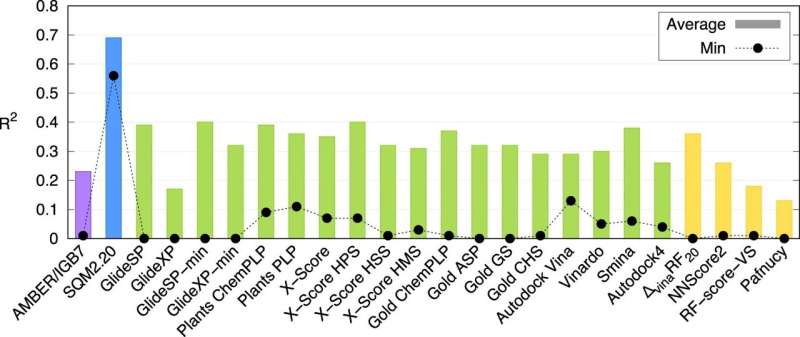
Studien visar att detta är den första universellt tillämpliga metoden i sitt slag. IOCB Prag beräkningsexperter testade det på 10 proteiner med olika nivåer av strukturell komplexitet, som var och en binder en stor mängd små molekyler (vanligtvis kallade ligander). De jämförde sedan sina resultat inte bara med andra motsvarande metoder, utan också med resultat från laboratorieexperiment, och båda jämförelserna visade sig vara mycket gynnsamma.
"Vi är naturligtvis inte de enda som arbetar med detta. Det finns flera sådana metoder. Vanligtvis kompenseras dock deras hastighet av låg noggrannhet medan mer exakta beräkningar kan ta flera dagar. Våra metoder är unika genom att de kan bearbeta information om stora molekylära system inom tiotals minuter samtidigt som man behåller fördelarna med mycket mer krävande kvantmekaniska beräkningar", förklarar Jan Řezáč, motsvarande författare till artikeln från gruppen Non-Covalent Interactions ledd av Prof. Pavel Hobza.
Experter från denna grupp har studerat intermolekylära interaktioner under lång tid. I denna forskning fokuserar de främst på biomolekyler, och resultaten av deras arbete har direkt betydelse för datorstödd design av läkemedel. Anledningen är att när forskare arbetar mot ett nytt läkemedel letar de ofta efter molekyler som binder starkt till ett visst protein.
Att identifiera dem är dock ungefär som att hitta nålar i en höstack, eftersom ett stort antal molekyler måste testas för att särskilja de som visar lovande. Detta fördröjer avsevärt upptäckten av medicinska substanser och gör det dyrare. Genom att förutsäga styrkan hos protein-ligandbindning, och på så sätt peka ut molekyler som bäst uppfyller en definierad uppsättning kriterier, skonar beräkningskemister arbetet från försöksledare, vilket i sin tur avsevärt påskyndar läkemedelsupptäckten.
Mer information: Adam Pecina et al, SQM2.20:Semiempirisk kvantmekanisk poängfunktion ger DFT-kvalitet protein-ligandbindningsaffinitetsförutsägelser på några minuter, Nature Communications (2024). DOI:10.1038/s41467-024-45431-8
Journalinformation: Nature Communications
Tillhandahålls av Institute of Organic Chemistry and Biochemistry of the CAS