Pepsi-SAXS:Ny metod för proteinanalys som är 50 gånger snabbare än analoger. Upphovsman:MIPT
Pepsi-SAXS är en ny, mycket effektiv metod för beräkning av röntgenspridningsprofiler, som behövs för analys av proteinmolekyl i lösningstillstånd. Metoden skapades av forskare från Université Grenoble Alpes och MIPT, ledd av Sergej Grudinin. Teamet testade sin metod, och resultaten publicerades av International Union of Crystallography i sin tidskrift Acta Crystallographica Avsnitt D:Strukturell biologi .
Proteiner har en komplex struktur och extremt liten storlek, i storleksordningen flera nanometer. För att studera dem, forskare måste hitta på ovanliga metoder, eftersom proteinprover alltför lätt förstörs och deras egenskaper ändras i experiment. Kunskap om strukturerna och funktionella mekanismerna för biomolekyler gör att nya läkemedel inte kan utvecklas genom försök och fel - tekniskt kallad screening för hög genomströmning - utan på ett mer fokuserat sätt.
En av de tekniker som används för att studera proteiner är analys av röntgenstrålar spridda från dem. Forskare behöver använda röntgenstrålar och inte vanligt ljus för att zooma in på enskilda atomer med en karakteristisk storlek i storleksordningen 0,1 nanometer. Ju mindre objektet är, desto kortare våglängd av ljus som måste användas för att observera det. Synligt ljus omfattar våglängder mellan 400 och 700 nanometer. Röntgen, å andra sidan, har en mycket kortare våglängd och kan därför användas för att undersöka molekylära strukturer.
"Den nya metoden gör att vi kan plotta spridningskurvor effektivt och exakt, och analysera den tredimensionella strukturen i ett prov, "säger MIPT -studenten Maria Garkavenko, medförfattare till tidningen. "Bland annat, Pepsi-SAXS ökar modelleringseffektiviteten och noggrannheten i tredimensionell makromolekylstrukturprognos. "
Småvinklad röntgenspridning, eller SAXS, är en experimentell teknik som innebär att röntgenstrålar sprids från ett prov och sedan samlas in i mycket små vinklar. Som ett resultat, en diagram över spridd röntgenstråleintensitet som funktion av infallsvinkeln erhålls. Med hjälp av denna tomt, ett proteinprov kan jämföras med andra prover i den experimentella databasen för att bestämma dess struktur och egenskaper.
Jämfört med andra tekniker som används för att bestämma provstruktur, SAXS är mycket enklare och billigare. Det kräver endast ett minimum av provberedning, och proteinerna behöver inte frysas eller kristalliseras. Proverna studeras i lösning och i deras funktionella tillstånd. Detta gör resultaten mycket mer tillförlitliga, eftersom provberedning ibland kan förändra tillståndet och egenskaperna hos ett protein. En annan viktig fördel med metoden är att den är icke-destruktiv, vilket innebär att det experimentella provet förblir i stort sett opåverkat av röntgenstrålar.
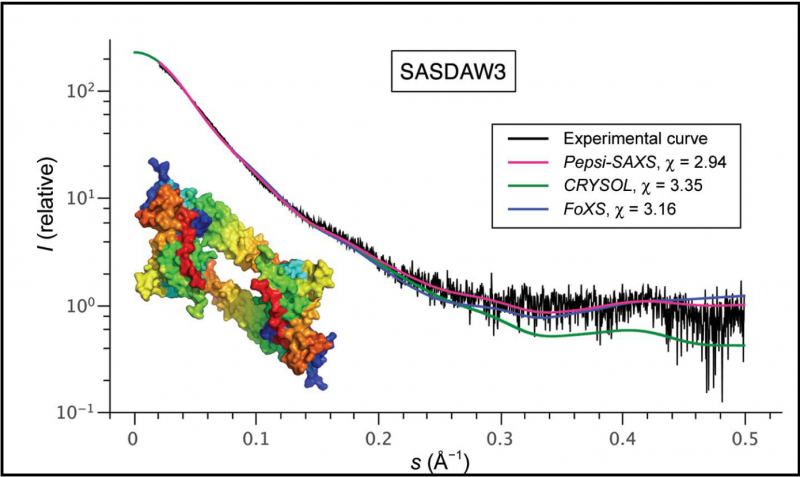
Figur 1 visar resultaten av en serie experiment, som jämförde Pepsi-SAXS med två av de för närvarande använda beräkningsmetoderna genom att tillämpa dem på samma prov (SASDAW3) från SASBDB-databasen. Genomsnittlig spridd intensitet ritas som en funktion av spridningsvinkeln. Felet ² i beräkningsmodellen är det lägsta när det gäller Pepsi-SAXS, vilket resulterar i en mer exakt bild av hydratiseringsskalet. Upphovsman:S. Grudinin, M. Garkavenko och A. Kazennov
Men tills nyligen, SAXS hade en stor nackdel:Metoden var beräkningsintensiv, vilket innebar att den inte kunde användas om antalet experiment var stort. Det tog timmar att bearbeta resultaten från bara ett experiment. Initialt, antalet beräkningar var direkt proportionellt mot kvadraten av antalet atomer i provet, det senare antalet brukar överstiga tusen. Dock, på 1970 -talet, Heinrich Stuhrmann, en tysk forskare, kom på en idé som förenklade beräkningarna. Han föreslog att spridning på molekylära föreningar skulle beskrivas i termer av funktioner av ett visst slag som kallas sfäriska övertoner. Detta tillvägagångssätt visade sig vara en framgång. Över åren, ett antal beräkningsverktyg för analys av SAXS -data skapades. Viktiga bidrag till deras utveckling gjordes av forskare med en sovjetisk vetenskaplig bakgrund inklusive Dmitri Svergun (arbetar för närvarande i Hamburg), som skrev programvarupaketet ATSAS för SAXS -dataanalys i biologisk makromolekylforskning. Forskarna i studien som rapporterats här undersökte flera beräkningsmetoder och jämförde dem med sin egen teknik.
"Pepsi-SAXS står för" polynomutvidgningar av proteinstrukturer och interaktioner "och" liten vinkel röntgenspridning. " Det är en adaptiv metod för snabb och exakt beräkning av småvinklade röntgenspridningsprofiler, "förklarar MIPT -doktoranden Andrei Kazennov, medförfattare till tidningen. "Pepsi-SAXS kan anpassas till storleken på ett givet prov och upplösningen av experimentella data."
Forskarna skapade också en effektiv modell av hydratiseringsskalet - ett lager av vattenmolekyler som omger proteiner i lösning - och införlivade det i sin programvara, öka metodens noggrannhet.
"Vår metod har validerats på en stor datamängd från BioIsis och SASBDB, de två största biologiska databaserna, säger Sergei Grudinin, som övervakade forskningen. "Vi har visat att Pepsi-SAXS är fem till 50 gånger snabbare än de tidigare använda metoderna, nämligen CRYSOL, FoXS, och den tredimensionella Zernike-tekniken implementerad i SAStbx-paketet. På samma gång, noggrannheten är i nivå med dem. "
Forskarna ägnade särskild uppmärksamhet åt analysen av de resultat de fick, som jämfördes med experimentella data.
Proteinforskning har grundläggande betydelse för vår förståelse av de grundläggande processerna som ligger till grund för livet, liksom för utveckling av läkemedel, behandlingar, och organiska material, inklusive konstgjorda organ. Det nya verktyget presenterat av författarna kan innebära 50 gånger snabbare framsteg inom dessa områden.