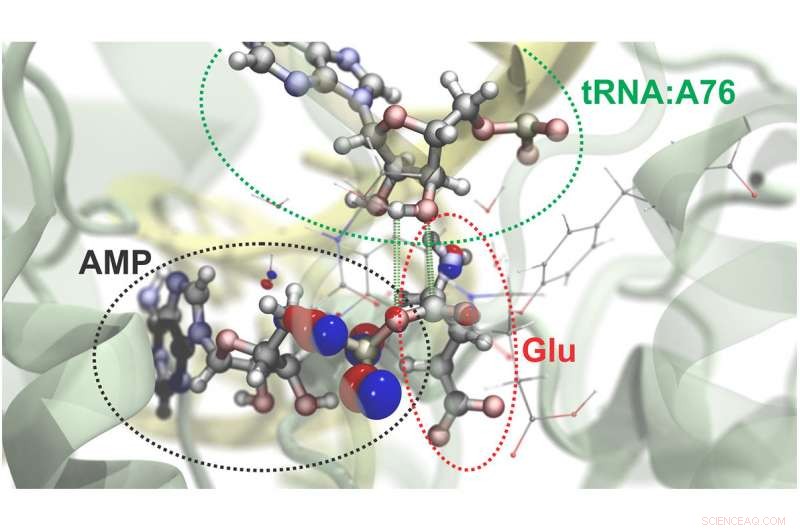
Forskare kan simulera atom- och subatomär dynamik i stora molekylära system. Här är en visualisering av processen genom vilken aminosyran glutamat (Glu) är bunden till en specifik region av dess överförings -RNA (tRNA). En energirik molekyl, ATP, driver denna reaktion och omvandlas till AMP i processen. De röda och blå bubblorna representerar sannolikheten att hitta elektroner i vissa regioner. Gröna prickade linjer avgränsar de atomer som binder i denna kemiska reaktion. Upphovsman:Rafael Bernardi, Zan Luthey-Schulten och Marcelo Melo
Forskare har byggt ett "beräkningsmikroskop" som kan simulera atom- och subatomära krafter som driver molekylära interaktioner. Detta verktyg kommer att effektivisera ansträngningarna för att förstå livets kemi, modellera stora molekylära system och utveckla nya farmaceutiska och industriella agenter, säger forskarna.
De rapporterar sina fynd i tidningen Naturmetoder .
Forskarna kombinerade två beräkningsmetoder som används för att simulera molekylära interaktioner. Den första, ett nanoskala molekylär-dynamikprogram som kallas NAMD, använder klassisk-mekaniska metoder för att modellera strukturen och simulera beteendet hos hundratals miljoner enskilda atomer. Det andra programmet zoomar in på det subatomära området, simulera interaktioner mellan protoner, neutroner och elektroner. Modellering i denna kvantmekaniska skala kräver mycket beräkningskraft, så implementerade forskarna en metod för att dela upp stora molekyler i klassiska och kvantmekaniska regioner. Detta gör att de kan fokusera sina beräkningsresurser på små regioner som är involverade i kritiska interaktioner, såsom att skapa eller bryta kemiska bindningar.
Både molekylärmekanik och kvantmekanikprogram har varit tillgängliga i åratal, och andra team har arbetat för att kombinera dem, sade University of Illinois kemiprofessor Zaida (Zan) Luthey-Schulten, som ledde den nya forskningen med sin man, U. av I. fysikprofessor Klaus Schulten. Men den nya ansträngningen effektiviserar processen med att skapa, utföra och analysera simuleringarna.
"Vi ställde in det så att forskare enkelt kan välja hur de ska dela upp sina egna system, "Sa Luthey-Schulten." Mina egna elever prövar det, och de flesta av dem kan göra det utan stora svårigheter. "
Schulten utvecklade NAMD i Illinois 1995, kombinerar den med en visualiseringsprogramvara, VMD, vilket gör det möjligt för forskare att se storskaliga molekylära interaktioner utvecklas. Schulten, som dog 2016, likställde detta tillvägagångssätt för att "bygga ett beräkningsmikroskop."
Beräkningsmikroskopet är idealiskt för modellering av strukturella drag och rörelser i stora komplex. Till exempel, under 2013, Schulten och hans kollegor använde NAMD för att modellera HIV -kapsiden, som består av mer än 1, 300 identiska proteiner som samlas i en cageliknande struktur som skyddar viruset tills det kommer in i en värdcell. Den simuleringen svarade för interaktionerna mellan mer än 64 miljoner atomer och krävde användning av Blue Waters superdator vid National Center for Supercomputing Applications i U. I. Den nya studien använde också Blue Waters, denna gång för att förbättra upplösningen av beräkningsmikroskopet.

Från vänster, doktorand Marcelo Melo, kemiprofessor Zaida Luthey-Schulten, postdoktoralforskaren Rafael Bernardi och deras kollegor har utvecklat ett nytt tillvägagångssätt för att modellera stora molekylära interaktioner på atom- och subatomära skalor. Deras arbete effektiviserar metoden för andra forskare och studenter. Upphovsman:L. Brian Stauffer
NAMD -programvaran är utformad för att beskriva beteendet hos enskilda atomer. Men enskilda atomer som är involverade i specifika kemiska interaktioner och reaktioner beter sig inte alltid som sina motsvarigheter någon annanstans. För att förstå hur de varierar krävs en närmare titt på de subatomära krafterna som spelar. Detta är särskilt viktigt i molekylernas dynamiska regioner - till exempel de platser där kemiska bindningar skapas eller bryts, sa forskarna.
I den nya studien, forskargruppen i Illinois samarbetade med QM -experter Frank Neese, från Max Planck Institute for Coal Research i Mulheim an der Ruhr, Tyskland; och Gerd B. Rocha, från Federal University of Paraiba, i Joao Pessoa, Brasilien.
Som en demonstration av det nya tillvägagångssättet, forskarna simulerade det kemiska beteendet hos överförings -RNA, molekyler som spelar en nyckelroll vid översättning av genetisk information till proteiner. Med NAMD, de modellerade den övergripande molekylstrukturen för tRNA just nu när ett speciellt protein laddar en aminosyra till tRNA. De delade upp två platser av komplexet i regioner som kräver ett mer fokuserat kvantmekaniskt tillvägagångssätt. (Se en film av simuleringen.)
De subatomära simuleringarna av interaktionerna mellan de två regionerna tillät laget att köra simuleringar av fyra olika scenarier som skulle tillåta tRNA att fungera som det gör i cellen. Deras simuleringar avslöjade att en av de fyra potentiella kemiska vägarna var mer energiskt gynnsam än de andra och därmed mer sannolikt att inträffa.
Forskarna använde också olika metoder för att dela tRNA -komplexet mellan MM- och QM -regionerna och rapporterade om varje tillvägagångssätt.
"Vi valde inte bara ett sätt, vi valde så många som möjligt. Vi ger användaren frihet. Hur du strukturerar det beror verkligen på vilket system du studerar, "sa U. av I. postdoktoralforskare Rafael Bernardi, en medförfattare i studien med doktoranden Marcelo Melo.
"Vi gör inte hela systemet kvantmekaniskt eftersom det skulle ta evigt att beräkna, Sa Melo.
"NAMD utformades - och det här var min mans vision - för att behandla riktigt stora system, "Luthey-Schulten sa." Nu kan vi lägga till den subatomära skalan till det, öppnar upp stora nya möjligheter för forskning. "