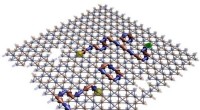
Installation av en adaptiv upplösningssimulering för fasta ämnen. Upphovsman:Springer
Datorsimuleringar används för att förstå egenskaperna hos mjuk materia - som vätskor, polymerer och biomolekyler som DNA -som är för komplicerade för att beskrivas med ekvationer. De är ofta för dyra för att simulera i sin helhet, med tanke på den intensiva beräkningskraft som krävs. Istället, en användbar strategi är att koppla ihop en exakt modell - tillämpad inom systemområden som kräver större uppmärksamhet - med en enklare, idealiserad modell.
I en ny tidning publicerad i EPJ E , Maziar Heidari, från Max Planck Institute for Polymer Research, Mainz, Tyskland och kollegor gör att den exakta modellen i högupplöst sammanfaller sömlöst med en exakt lösbar representation vid lägre upplösning.
Idealet, enklare modell är en slags naken representation av atomer eller molekyler, som inte interagerar med varandra. Tidigare studier har tillämpat denna strategi på vätskor, men i denna studie, författarna tillämpar den för första gången på en modellfast kopplad till en idealisk kristall, där atomer har begränsade rörelser och inte interagerar, kallad Einstein -kristall. Teamet kunde beräkna sina termodynamiska egenskaper - t.ex. temperatur och fri energi - till en lägre beräkningskostnad.
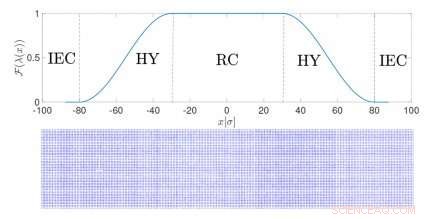
I denna typ av simulering, kallas adaptiv upplösningssimuleringar, upplösningen av en molekyl beror på dess position i rymden. I övergångsregionen mellan de två resolutionerna, molekyler anpassar sig till den ena modellen eller den andra. Detta är ett effektivt sätt att beräkna de relevanta termodynamiska egenskaperna hos det faktiska fasta materialet genom att sönderdela dem i ett idealiskt bidrag - från den förenklade modellen - och en annan term, specifikt för det specifika systemet. Metoden kombinerar enkelheten hos ideala modeller med den kemiska noggrannheten hos realistiska representationer.