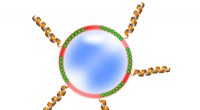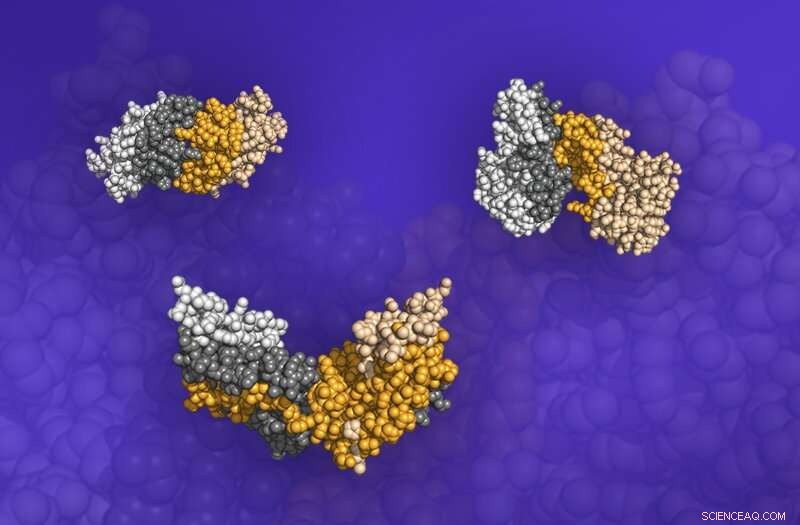
Tre dimerer, proteinstrukturer bestående av två bundna proteiner, från Dockground -databasen. Gränssnitten vid vilka proteinerna möts visas som de mörkade områdena. Kredit:ORNL
Kan något så enkelt som form helt avgöra om proteiner kommer att binda ihop eller inte? Forskare ger superdatorer i uppdrag att ta reda på det.
Ett team under ledning av Sharon Glotzer, framstående professor och avdelningsordförande i kemiteknik vid University of Michigan (UM), använde 200-petaflop Summit superdator vid US Department of Energy (DOE:s) Oak Ridge National Laboratory (ORNL) för att modellera lock-and-key interaktioner mellan proteiner för att studera deras bindande beteenden. Resultaten, publicerad i Mjuk materia, avslöjade att vissa proteiner gör, faktiskt, binda baserat på enbart form.
"Vi har visat att något så enkelt som form kan förutsäga proteininteraktioner som ibland är riktigt komplexa, "sa Jens Glaser, beräkningsvetare i gruppen Advanced Computing for Chemistry and Materials vid Oak Ridge Leadership Computing Facility (OLCF). "Denna första demonstration har fått oss att tro att formen har varit en ouppskattad ingrediens i många proteinmonteringsprocesser."
Resultaten kan ha många tillämpningar inom biologisk forskning. Till exempel, metoden kan användas för att screena läkemedel mot sjukdomar eller ge forskare information om hur man använder proteiner som byggstenar för att designa nya biologiska material.
"Denna spännande studie visar kraften i formkomplementaritet i förutsägelsen av protein-protein-gränssnitt, "sa doktor Stephanie McElhinny, programchef vid US Army Combat Capabilities Development Command's Army Research Laboratory, med hänvisning till det gynnsamma rumsliga förhållandet mellan två kompatibelt formade proteiner. "Beräkningsmodeller som exakt förutsäger dessa gränssnitt kommer att stödja framtida design av avancerade proteinbaserade material med aktiva och responsiva egenskaper, till exempel lättskördad proteinbaserad plast som kan fungera som ett konstgjort blad för kraftproduktion. "
Superdatorer avslöjar att form är nyckeln till vissa proteiner
För att proteiner med framgång ska binda till varandra, en av dem fungerar som en ligand, en molekyl som fäster vid ett målprotein, och en av dem fungerar som en receptor, molekylen som tar emot liganden. Denna process involverar komplexa kemiska interaktioner, i vilka molekyler delar bindningar och ändrar sina konfigurationer vid bindning.
Glotzers team ville se om de kunde förutsäga denna molekylära bindning baserat på enbart form, ignorerar interaktionen mellan proteiner. Från en databas med mer än 6, 000 proteinpar, laget testade 46 par som är kända för att binda till varandra och simulerade deras sammansättning på toppmötet. Teamet utförde simuleringarna under programmet INCITE (Innovative and Novel Computational Impact on Theory and Experiment).
Som att flera tennisbollar kastas mot ett enda mål, simuleringarna modellerade flera ligander som kastas på en enda, fixerad målreceptor. Av de 46 testade paren de hittade 6 par som presterade bra - mer än 50 procent av tiden de lyckades montera baserat enbart på deras kompletterande former.
"Vi tittade på gränssnitten där proteinerna band ihop för att se hur lika de var i deras verkliga gränssnitt, och sedan bestämde vi avstängningen för att se hur många par som var goda förutsägare för de verkliga gränssnitten, "sa Fengyi Gao, Ph.D. kandidat vid UM. "Vi fann att 13 procent av dessa proteinpar kunde binda baserat på enbart form."
Teamet byggde sedan en maskininlärningsmodell som kunde avgöra vilka proteiner som kan monteras enbart baserat på form. Att kombinera sin initiala modell med sådana maskininlärningsverktyg hjälper dem att förstå vilken information som behövs för proteinpar som inte kan monteras baserat på enbart formkomplementaritet.
Kör proteiner parallellt
För att modellera flera reversibla bindningsprocesser för 46 proteinpar under olika parametrar, de behövde två dagars beräkningstid och mer än 3, 000 grafikkort - en summa som bara en superdator som OLCF:s toppmöte skulle kunna ge. OLCF är ett DOE Office of Science User Facility på ORNL.
Som en del av HOOMD-blå beräkningskod som användes för att köra simuleringarna, Glaser, som tidigare var assisterande forskare i Glotzers grupp vid UM, utvecklat en algoritm som simulerade proteinerna i närvaro av många små partiklar. Men Glaser hittade ett sätt att bara modellera rörelsen för de proteiner som laget var intresserad av, undvika onödiga och dyra beräkningar för lösningsmedelsmolekylerna runt dem.
"Jag körde koden parallellt så att många olika parametrar, iterationer av samma system, och olika proteiner kan distribueras över GPU:erna, "Sa Glaser." Detta gjorde det möjligt för oss att enkelt utnyttja Summits parallella datormöjligheter. "
Med Summit, laget fångade sex proteinpar som bundit endast baserat på formkomplementaritet, med en av dem som uppnår bindning mer än 94 procent av tiden.
"Det var ganska förvånande för oss att en sådan förenklad modell korrekt kunde välja just den ena posen som de antar av de många hundra eller fler poserna som tävlar, "Glaser sa." Vi förväntade oss att mycket mer skulle vara nödvändigt för att reproducera den verkliga bindningsställningen för dessa proteinpar. "
Modeller kan hjälpa till med läkemedelsscreening
Teamet planerar att studera fler proteiner som också kan binda baserat på form - eller bilda ännu högre ordningsstrukturer. Teamets nuvarande studie undersökte endast proteindimerer, som består av två proteiner bundna tillsammans, men teamet vill veta begränsningen för hur proteinformer kan utvecklas för att bilda hierarkiska proteinstrukturer.
"Innan vi gjorde denna studie, Jag trodde faktiskt inte att proteiner kunde bilda dimerer baserat på enbart form, "sa Fengyi Gao, Ph.D. kandidat vid UM. "Men nu, vi har funnit att det här fungerar, och vi kan studera mer komplexa strukturer eller till och med kombinera detta med andra tillvägagångssätt, som maskininlärning, för att se vilka funktioner vi behöver för att möjliggöra korrekt bindning. "
Teamet hoppas att de så småningom kan förutsäga bindningen av protein-protein-gränssnitt i proteinkluster eller proteinkristallisationsstrukturer.
"Vi tror att vi kan anpassa detta tillvägagångssätt till något som läkemedelsscreening i framtiden, "Sa Gao." Utöver det, Vi hoppas att den här formbaserade modellen kan fungera som en grund för att studera proteinmontering i allmänhet. "