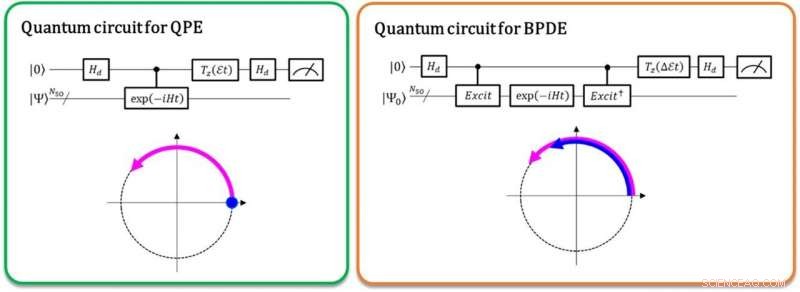
Vänster:Fasskillnaden mellan | 0⟩ | Ψ⟩ och exp (-iEt) | 1⟩ | Ψ⟩ ger den totala energin E. Den krökta pilen i lila indikerar fasutvecklingen av | Ψ⟩ i tid. Höger:Fasskillnaden mellan exp (-iE0t) | 0⟩ | Ψ0⟩ och exp (-iE1t) | 1⟩ | Ψ1⟩ ger energiskillnaden E1-E0, direkt. De krökta pilarna i blått och lila indikerar fasutvecklingen för | Ψ0⟩ och den för | Ψ1⟩, respektive. Upphovsman:K. Sugisaki, K. Sato och T. Takui
Som nyligen rapporterats av tidningen Fysisk kemi Kemisk fysik , forskare från Graduate School of Science vid Osaka City University har utvecklat en kvantalgoritm som kan förstå de elektroniska tillstånden i atom- eller molekylsystem genom att direkt beräkna energiskillnaden i deras relevanta tillstånd. Implementerad som en Bayesiansk fasuppskattning, algoritmen bryter från konventionen genom att inte fokusera på skillnaden i totala energier beräknade från utvecklingen före och efter fas, men genom att följa utvecklingen av själva energiskillnaden.
"Nästan alla kemiproblem diskuterar energiskillnaden, inte molekylens totala energi, "säger forskningsansvarig och särskilt utsedd föreläsare Kenji Sugisaki, "också, molekyler med tunga atomer som förekommer i den nedre delen av det periodiska systemet har stora totala energier, men storleken på energiskillnaden som diskuteras i kemi, såsom elektroniska excitationstillstånd och joniseringsenergier, beror inte mycket på molekylens storlek. "Denna idé ledde Sugisaki och hans team till att implementera en kvantalgoritm som direkt beräknar energiskillnader istället för totala energier, skapa en framtid där skalbara eller praktiska kvantdatorer gör att vi kan utföra verklig kemisk forskning och materialutveckling.
För närvarande, kvantdatorer kan utföra beräkningarna med fullständig konfigurationsinteraktion (full-CI) som ger optimala molekylära energier med en kvantalgoritm som kallas kvantfasestimering (QPE), noterar att full-CI-beräkningen för betydande molekylära system är svårhanterlig med alla superdatorer. QPE förlitar sig på det faktum att en vågfunktion, | Ψ⟩ som betecknar den matematiska beskrivningen av ett mikroskopiskt systems kvanttillstånd-i det här fallet den matematiska lösningen av Schrödinger-ekvationen för det mikroskopiska systemet, såsom en atom eller molekyl-ändrar tids-evolutionärt sin fas beroende på dess totala energi. I den konventionella QPE, kvantsuperpositionstillståndet (| 0⟩ | Ψ⟩+| 1⟩ | Ψ⟩) ⁄ √2 bereds, och införandet av en kontrollerad tidsutvecklingsoperatör får | Ψ⟩ bara att utvecklas i tid när den första qubit anger tillståndet | 1⟩. Således, tillståndet | 1⟩ skapar en kvantfas av efterutvecklingen i tid medan | 0⟩ anger förutvecklingens. Fasskillnaden mellan för- och efterutvecklingen ger systemets totala energi.
Forskarna vid Osaka City University generaliserar den konventionella QPE till den direkta beräkningen av skillnaden i den totala energin mellan två relevanta kvanttillstånd. I den nyligen implementerade kvantalgoritmen benämnd Bayesian fasdifferensestimering (BPDE), överlagringen av de två vågfunktionerna, (| 0⟩ | Ψ 0 ⟩ + | 1⟩ | Ψ 1 ⟩) ⁄ √2, var | Ψ 0 ⟩ Och | Ψ 1 ⟩ Beteckna vågfunktionen som är relevant för varje tillstånd, respektive, är förberedd, och skillnaden i fasen mellan | Ψ 0 ⟩ Och | Ψ 1 ⟩ Efter den tid som evolutionen av superpositionen ger direkt skillnaden i den totala energin mellan de två inblandade vågfunktionerna. "Vi betonar att algoritmen följer utvecklingen av energiskillnaden över tid, som är mindre benägen för buller än att individuellt beräkna den totala energin för en atom eller molekyl. Således, algoritmen passar behovet av kemiproblem som kräver exakt noggrannhet i energi. "säger forskarhandledare och professor emeritus Takeji Takui.
Tidigare, denna forskargrupp utvecklade en kvantalgoritm som direkt beräknar energiskillnaden mellan elektroniska tillstånd (spinntillstånd) med olika spinnkvantnummer (K. Sugisaki, K. Toyota, K. Sato, D. Shiomi, T. Takui, Chem. Sci. 2021, 12 , 2121–2132.). Denna algoritm, dock, kräver fler qubits än den konventionella QPE och kan inte tillämpas på energidifferensberäkningen mellan de elektroniska tillstånden med lika snurrande kvantnummer, vilket är viktigt för spektral tilldelning av UV-synliga absorptionsspektra. BPDE -algoritmen som utvecklats i studien övervinner dessa frågor, vilket gör det till en mycket mångsidig kvantalgoritm.