Materialforskare och ingenjörer skulle vilja veta exakt hur elektroner interagerar och rör sig i nya material och hur de enheter som tillverkas med dem kommer att bete sig. Kommer den elektriska strömmen att flyta lätt i materialet? Finns det en temperatur vid vilken materialet blir supraledande, vilket gör att ström kan flyta utan strömkälla? Hur länge kommer ett elektronspins kvanttillstånd att bevaras i nya elektroniska och kvantenheter?
En gemenskap av materialfysiker försöker ta itu med sådana frågor genom att förstå vad som sker inuti material, beräkna deras beteende ner till nivån av individuella elektroninteraktioner och atomrörelser.
Nu har ett Caltech-team gjort en nyckelupptäckt som hjälper till att förenkla sådana beräkningar, påskynda dem med en faktor på 50 eller mer samtidigt som noggrannheten bibehålls. Som ett resultat är det möjligt att beräkna elektroninteraktioner i mer komplexa material och enheter samt att utveckla nya beräkningar som tidigare ansågs omöjliga.
I en ny artikel publicerad i tidskriften Physical Review X , Caltechs Yao Luo, en doktorand i tillämpad fysik; hans rådgivare Marco Bernardi, professor i tillämpad fysik, fysik och materialvetenskap; och kollegor beskriver en ny datadriven metod som har möjliggjort dessa framsteg. Deras tillvägagångssätt förenklar de täta beräkningsmatriser som används för att representera de interaktioner som äger rum i ett material mellan elektroner och atomvibrationer (eller fononer, som kan ses som individuella enheter av vibrationsenergi).
Luo och Bernardi säger att den nya metoden tillåter dem att använda endast 1 till 2 % av den data som vanligtvis används för att lösa sådana problem, vilket avsevärt accelererar beräkningar och i processen avslöjar de viktigaste interaktionerna som dikterar materialens egenskaper.
"Det här var väldigt överraskande", säger Bernardi. "Elektron-fonon-interaktionerna som beräknas med de komprimerade matriserna är nästan lika exakta som hela beräkningen. Detta minskar beräkningstiden och minnesanvändningen enormt, med ungefär två storleksordningar i de flesta fall. Det är också ett elegant exempel på Occams rakhyvel, idén att gynna enkla fysiska modeller med minimalt antal parametrar."
Forskare inom detta område följer i allmänhet en av två metoder för att förstå material på denna mest grundläggande nivå. Ett tillvägagångssätt betonar att bygga minimala modeller, vilket minskar komplexiteten i systemet, så att forskare kan justera en handfull parametrar i penna-och-papper-beräkningar för att få en kvalitativ förståelse av material.
Den andra börjar med ingenting annat än strukturen hos ett material och använder så kallade "första principer"-metoder – kvantmekaniska beräkningar som kräver stora datorer – för att studera materialegenskaper med kvantitativ noggrannhet.
Denna senare uppsättning metoder, som Bernardis grupp fokuserar på, använder extremt stora matriser med miljarder poster för att beräkna elektroninteraktioner som styr ett brett spektrum av fysikaliska egenskaper. Det översätts till tusentals timmars beräkningstid för varje beräkning. Det nya verket föreslår ett slags mellanväg mellan de två tillvägagångssätten, säger Bernardi.
"Med vår nya metod kan du trunkera storleken på dessa matriser, extrahera nyckelinformationen och generera minimala modeller av interaktioner i material."
Hans grupps tillvägagångssätt bygger på att tillämpa en metod som kallas singular value decomposition (SVD) på elektron-fonon-interaktionerna i ett material. SVD-tekniken används ofta inom områden som bildkomprimering och kvantinformationsvetenskap. Här gör det det möjligt för författarna att separera, eller isär, de elektroniska och vibrationskomponenterna i en matris av tusentals eller miljoner elektron-fonon-interaktioner och att tilldela varje grundläggande interaktion ett nummer.
Dessa reella positiva siffror kallas singulära värden och rangordnar de grundläggande interaktionerna efter betydelse. Då kan programmet eliminera alla utom några få procent av interaktionerna i varje matris, och bara lämna kvar de ledande singularvärdena, en process som gör bestämningen billigare med en faktor som är proportionell mot mängden komprimering.
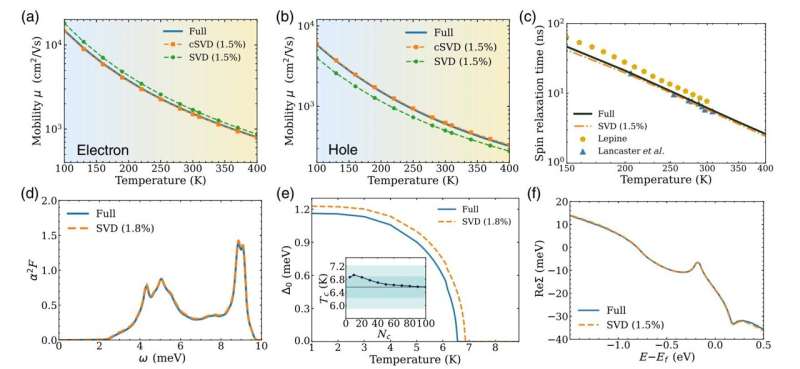
Så, till exempel, om programmet bara behåller 1 % av singularvärdena, blir beräkningen snabbare med en faktor 100. Forskarna har funnit att om man bara behåller en liten del av singularvärdena, vanligtvis 1 till 2 %, blir det ungefärliga resultatet behåller nästan samma noggrannhet som hela beräkningen.
"Genom att använda SVD kan du minska antalet singularvärden och bara fånga huvuddragen i matriserna som representerar elektroniska interaktioner i ett givet material", säger Luo, huvudförfattare på tidningen som går tredje året i Bernardis grupp.
"Detta trunkerar den ursprungliga matrisen, vilket påskyndar algoritmen och har den extra fördelen att avslöja vilka interaktioner i materialet som är dominerande."
Bernardi noterar att denna senare fördel med SVD-metoden ger forskarna en "fysisk intuition" om elektroninteraktioner i ett material, något som har saknats från de första principberäkningarna tidigare. Till exempel, i en beräkning som involverade kisel, blev det tydligt att det dominerande singularvärdet var associerat med sträckningen och klämningen av en viss bindning.
"Det är något enkelt, men innan vi gjorde beräkningen visste vi inte att det var den starkaste interaktionen", förklarar Bernardi.
I uppsatsen visar forskarna att komprimeringen av matriser relaterade till elektron-fonon-interaktioner med SVD-metoden ger korrekta resultat för olika egenskaper hos material som forskare kanske vill beräkna, inklusive laddningstransport, spinrelaxationstider och övergångstemperaturen för supraledare .
Bernardi och hans team utökar de SVD-baserade beräkningarna till ett bredare utbud av interaktioner i material och utvecklar avancerade beräkningar som tidigare ansågs omöjliga. Teamet arbetar också med att lägga till den nya SVD-metoden i sin open source Perturbo-kod, ett mjukvarupaket som hjälper forskare att beräkna hur elektroner interagerar och rör sig i material. Bernardi säger att detta kommer att göra det möjligt för användare i forskarvärlden att förutsäga materialegenskaper associerade med elektron-fonon-interaktioner betydligt snabbare.
Uppsatsen har titeln "Datadriven komprimering av elektron-fonon-interaktioner." Tillsammans med Luo och Bernardi, medförfattare på tidningen inkluderar doktoranden Dhruv Desai (MS '22); Benjamin Chang (MS '20) och Jinsoo Park (Ph.D. '22), som nu är postdoktor vid University of Chicago.