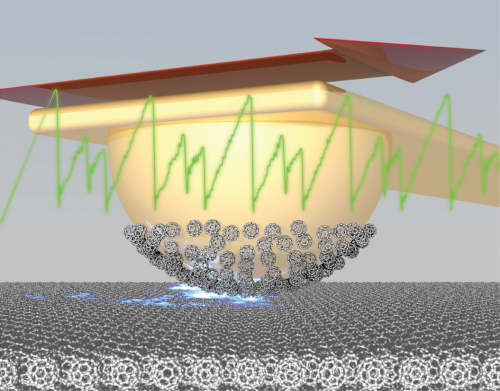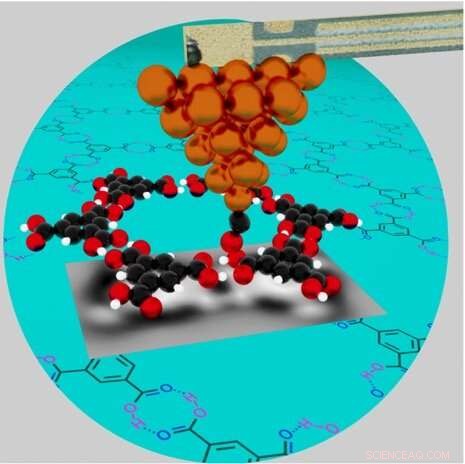
En illustration av ett högupplöst atomkraftsmikroskop som undersöker de kemiska egenskaperna hos vätebunden trimesinsyra (TMA) nätverk (överlagrat på en kricka cirkel) på en kopparyta. Nyckel:kopparatomer på metallspetsens spets (orange), kolatomer (svart), syreatomer (röda) och väteatomer (vita). Den enda kolmonoxid (CO) molekylen i slutet av spetsen spets, med kolet fäst till koppar, är lite böjd som svar på de frånstötande krafterna från det närliggande syret i TMA molekylen. Kredit:Brookhaven National Laboratory
Bläddra igenom en lärobok i kemi och du kommer att se ritningar av den kemiska strukturen hos molekyler – där enskilda atomer är ordnade i rymden och hur de är kemiskt bundna till varandra. I årtionden kunde kemister bara indirekt bestämma kemiska strukturer baserat på responsen som genererades när prover interagerade med röntgenstrålar eller ljuspartiklar. För det speciella fallet med molekyler på en yta, gav atomkraftsmikroskopi (AFM), som uppfanns på 1980-talet, direkta bilder av molekyler och de mönster de bildar när de sätts samman till tvådimensionella (2D) arrayer. Under 2009 gjorde betydande framsteg inom högupplöst AFM (HR-AFM) det möjligt för kemister för första gången att direkt avbilda den kemiska strukturen hos en enskild molekyl med tillräckliga detaljer för att särskilja olika typer av bindning inuti molekylen.
AFM "känner" krafterna mellan en vass sondspets och ytatomer eller molekyler. Spetsen skannar över en provyta, från vänster till höger och uppifrån och ned, på en höjd av mindre än en nanometer, och registrerar kraften vid varje position. En dator kombinerar dessa mätningar för att generera en kraftkarta, vilket resulterar i en ögonblicksbild av ytan. AFM finns i laboratorier över hela världen och är arbetshästinstrument med olika tillämpningar inom vetenskap och teknik.
Endast ett fåtal HR-AFM finns i USA. Den ena är belägen vid Center for Functional Nanomaterials (CFN)—en US Department of Energy (DOE) Office of Science User Facility vid Brookhaven National Laboratory. I flera år har fysikern Percy Zahl från CFN Interface Science and Catalysis Group uppgraderat och anpassat CFN HR-AFMs hårdvara och mjukvara, vilket gör det lättare att använda och skaffa bilder. Som mycket specialiserade instrument kräver HR-AFM expertis att använda. De fungerar vid mycket låg temperatur (precis över den som krävs för att göra helium flytande). Dessutom beror HR-avbildning på att fånga en enda kolmonoxidmolekyl i änden av spetsen.
Lika utmanande som att förbereda och använda instrumentet för experiment kan vara, att se hur molekyler ser ut är bara början. Därefter måste bilderna analyseras och tolkas. Med andra ord, hur korrelerar bildegenskaper med molekylers kemiska egenskaper?
Tillsammans med teoretiker från CFN och universitet i Spanien och Schweiz ställde Zahl just denna fråga för vätebundna nätverk av trimesinsyra (TMA) molekyler på en kopparyta. Zahl började avbilda dessa porösa nätverk — gjorda av kol, väte och syre — för några år sedan. Han var intresserad av deras potential att begränsa atomer eller molekyler som kan vara värd för elektronspintillstånd för kvantinformationsvetenskap (QIS) tillämpningar. Men enbart med experiment och grundläggande simuleringar kunde han inte förklara deras grundläggande struktur i detalj.
"Jag misstänkte att TMA-molekylernas starka polaritet (laddningsområden) låg bakom det jag såg på AFM-bilderna", sa Zahl. "Men jag behövde mer exakta beräkningar för att vara säker."
I AFM mäts den totala kraften mellan sondspetsen och molekylen. Men för en exakt matchning mellan experiment och simulering måste varje enskild kraft på spel redovisas. Grundmodeller kan simulera kortdistanskrafter för enkla opolära molekyler, där elektriska laddningar är jämnt fördelade. Men för kemiskt rika strukturer som finns i polära molekyler som trimesinsyra måste elektrostatiska krafter (som härrör från den elektroniska laddningsfördelningen inuti molekylen) och van der Waals-krafter (attraktion mellan molekyler) också beaktas. För att simulera dessa krafter behöver forskare den exakta molekylgeometrin som visar hur atomer är placerade i alla tre dimensionerna och de exakta laddningsfördelningarna inuti molekylerna. Genom DFT-beräkningar vid Swiss National Supercomputing Center, slappnade Aliaksandr Yakutovich strukturellt av ringen med sex TMA-molekyler på en kopparplatta innehållande 1 800 kopparatomer. Vid strukturell relaxation optimeras en grundläggande geometrisk eller strukturell modell för att hitta konfigurationen av atomer med lägsta möjliga energi.
I denna studie analyserade Zahl arten av självmontering av TMA-molekyler till bikakeliknande nätverksstrukturer på en ren kopparkristall. Zahl avbildade först strukturerna i stor skala med ett scanning tunneling microscope (STM). Detta mikroskop skannar en metallspets över en yta samtidigt som en elektrisk spänning appliceras mellan dem. För att identifiera hur nätverksstrukturen var i linje med substratet bombarderade CFN-materialforskaren Jurek Sadowski provet med lågenergielektroner och analyserade mönstret av diffrakterade elektroner. Slutligen utförde Zahl HR-AFM, som är känslig för höjden av ytegenskaper på en submolekylär skala.
"Med STM kan vi se nätverken av TMA-molekyler men kan inte lätt se orienteringen av koppar samtidigt", sa Zahl. "Lågenergielektrondiffraktion kan berätta för oss hur koppar- och TMA-molekylerna är orienterade i förhållande till varandra. AFM tillåter oss att se den detaljerade kemiska strukturen hos molekylerna. Men för att förstå dessa detaljer måste vi modellera systemet och bestämma exakt där TMA-molekylernas atomer sitter på koppar."
För denna modellering använde teamet densitetsfunktionella teorin (DFT) för att beräkna de mest energimässigt gynnsamma arrangemangen av TMA-molekyler på koppar. Tanken bakom DFT är att den totala energin i ett system är en funktion av dess elektrondensitet, eller sannolikheten att hitta en elektron på en viss plats runt en atom. Fler elektronegativa atomer (som syre) tenderar att dra elektroner bort från mindre elektronegativa atomer (som kol och väte) de är bundna till, liknande en magnet. Sådana elektrostatiska interaktioner är viktiga för att förstå kemisk reaktivitet.
Mark Hybertsen, ledare för CFN Theory and Computation Group, utförde initiala DFT-beräkningar för en individuell TMA-molekyl och två TMA-molekyler sammanfogade av vätebindningar (en dimer). Aliaksandr Yakutovich från [email protected] Laboratory of the Swiss Federal Laboratories for Materials Science and Technology (Empa) körde sedan DFT-beräkningar av ett större TMA-nätverk som består av en komplett ring av sex TMA-molekyler.
Dessa beräkningar visade hur molekylernas inre kolring förvrängs från en hexagonal till en triangulär form i AFM-bilden på grund av starka polarisationer orsakade av tre karboxylgrupper (COOH). Dessutom dras eventuella obundna syreatomer en bit ner mot ytan av kopparatomer, där fler elektroner finns. De beräknade också styrkan hos de två vätebindningarna som bildas mellan två TMA-molekyler. Dessa beräkningar visade att varje bindning var ungefär dubbelt så stark som en typisk enkelvätebindning.
"Genom att koppla modeller i atomskala till AFM-avbildningsexperimenten kan vi förstå grundläggande kemiska egenskaper i bilderna", säger Hybertsen.
"Denna förmåga kan hjälpa oss att identifiera kritiska molekylegenskaper, inklusive reaktivitet och stabilitet, i komplexa blandningar (som petroleum) baserade på HR-AFM-bilder," tillade Zahl.

En jämförelse mellan experimentella (överst) och simulerade (nedersta tre vid olika höjder av probspetsprov) AFM-bilder av två vätebundna TMA-molekyler. Kredit:Brookhaven National Laboratory
För att sluta slingan mellan modellering och experiment matade samarbetspartners i Spanien in DFT-resultaten i en beräkningskod som de utvecklade för att generera simulerade AFM-bilder. Dessa bilder matchade perfekt de experimentella.
"Dessa exakta simuleringar avslöjar det subtila samspelet mellan den ursprungliga molekylstrukturen, deformationer som induceras av interaktionen med substratet och de inneboende kemiska egenskaperna hos molekylen som bestämmer komplexet, slående kontrast som vi observerar i AFM-bilderna", säger Ruben Perez från Universidad Autónoma de Madrid.
Från deras kombinerade tillvägagångssätt visade teamet också att linjeliknande egenskaper som förekommer mellan molekyler i AFM-bilder av TMA (och andra molekyler) inte är fingeravtryck av vätebindningar. Snarare är de "artefakter" från böjning av AFM-sondmolekylen.
"Även om vätebindning är mycket stark för TMA-molekyler, är vätebindningar osynliga i experimentet och simuleringen," sa Zahl. "Det som är synligt är bevis på stark elektronborttagning av karboxylgrupperna."
Därefter planerar Zahl att fortsätta studera detta modellsystem för självmontering av nätverk för att utforska dess potential för QIS-applikationer. Han kommer att använda ett nytt STM/AFM-mikroskop med ytterligare spektroskopiska möjligheter, såsom de för att kontrollera prover med ett magnetfält och applicera radiofrekventa fält på prover och karakterisera deras svar. Dessa funktioner gör det möjligt för Zahl att mäta kvantspinntillstånden för anpassade molekyler arrangerade i en perfekt array för att bilda potentiella kvantbitar.
Forskningen publicerades i Nanoscale . + Utforska vidare