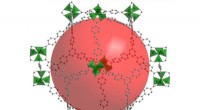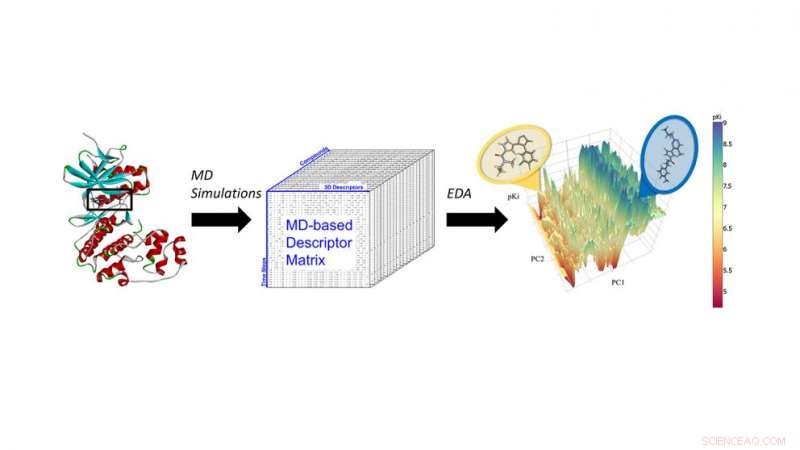
Molecular dynamics (MD) simuleringar av ERK2-hämmare för att extrahera MD-deskriptorer för nästa generations keminformatikanalys och maskininlärning. Kredit:North Carolina State University
Forskare från North Carolina State University har visat att simuleringar av molekylär dynamik och maskininlärningstekniker kan integreras för att skapa mer exakta datorförutsägelsemodeller. Dessa "hyperprediktiva" modeller kan användas för att snabbt förutsäga vilka nya kemiska föreningar som kan vara lovande läkemedelskandidater.
Läkemedelsutveckling är en kostsam och tidskrävande process. För att begränsa antalet kemiska föreningar som kan vara potentiella läkemedelskandidater, forskare använder datormodeller som kan förutsäga hur en viss kemisk förening kan interagera med ett biologiskt mål av intresse - till exempel, ett nyckelprotein som kan vara involverat i en sjukdomsprocess. Traditionellt, detta görs via kvantitativ struktur-aktivitetsrelation (QSAR) modellering och molekylär dockning, som förlitar sig på 2- och 3D-information om dessa kemikalier.
Denis Fourches, biträdande professor i beräkningskemi, ville förbättra noggrannheten hos dessa QSAR -modeller. "När du screenar en uppsättning av 30 miljoner föreningar, du behöver inte nödvändigtvis ha en mycket hög tillförlitlighet med din modell - du får bara en idé om de fem eller tio procent av det virtuella biblioteket. Men om du försöker begränsa ett fält med 200 analoger till 10, vilket är vanligare vid läkemedelsutveckling, din modelleringsteknik måste vara extremt exakt. Nuvarande tekniker är definitivt inte tillräckligt tillförlitliga."
Fourches och Jeremy Ash, en doktorand i bioinformatik, beslutat att införliva resultaten av molekylära dynamikberäkningar - simuleringar av alla atomer av hur en viss förening rör sig i bindningsfickan på ett protein - i förutsägelsemodeller baserade på maskininlärning.
"De flesta modeller använder bara de tvådimensionella strukturerna av molekyler, " säger Fourches. "Men i verkligheten, kemikalier är komplexa tredimensionella föremål som rör sig, vibrerar och har dynamiska intermolekylära interaktioner med proteinet när det väl har dockats i dess bindningsställe. Du kan inte se det om du bara tittar på 2D- eller 3D-strukturen för en given molekyl."
I en proof-of-concept-studie, Fourches och Ash tittade på ERK2-kinaset - ett enzym associerat med flera typer av cancer - och en grupp av 87 kända ERK2-hämmare, allt från mycket aktiv till inaktiv. De körde oberoende simuleringar av molekylär dynamik (MD) för var och en av dessa 87 föreningar och beräknade kritisk information om flexibiliteten för varje förening en gång i ERK2-fickan. Sedan analyserade de MD-deskriptorerna med hjälp av keminformatiktekniker och maskininlärning. MD-deskriptorerna kunde exakt skilja aktiva ERK2-hämmare från svagt aktiva och inaktiva, vilket inte var fallet när modellerna endast använde 2-D och 3-D strukturell information.
"Vi hade redan data om dessa 87 molekyler och deras aktivitet vid ERK2, ”, säger Fourches. ”Så vi testade för att se om vår modell kunde hitta de mest aktiva föreningarna på ett tillförlitligt sätt. Verkligen, den skilde exakt mellan starka och svaga ERK2-hämmare, och eftersom MD -deskriptorer kodade för interaktioner som föreningarna skapar i fickan på ERK2, det gav oss också mer inblick i varför de starka hämmarna fungerade bra.
"Innan datorframsteg tillät oss att simulera den här typen av data, det skulle ha tagit oss sex månader att simulera en enda molekyl i fickan på ERK2. Tack vare GPU-acceleration, nu tar det bara tre timmar. Det är en game changer. Jag är hoppfull att inkorporering av data extraherad från molekylär dynamik i QSAR-modeller kommer att möjliggöra en ny generation av hyperprediktiva modeller som hjälper till att skapa nya, effektiva läkemedel på marknaden ännu snabbare. Det är artificiell intelligens som arbetar för oss att upptäcka morgondagens droger. "