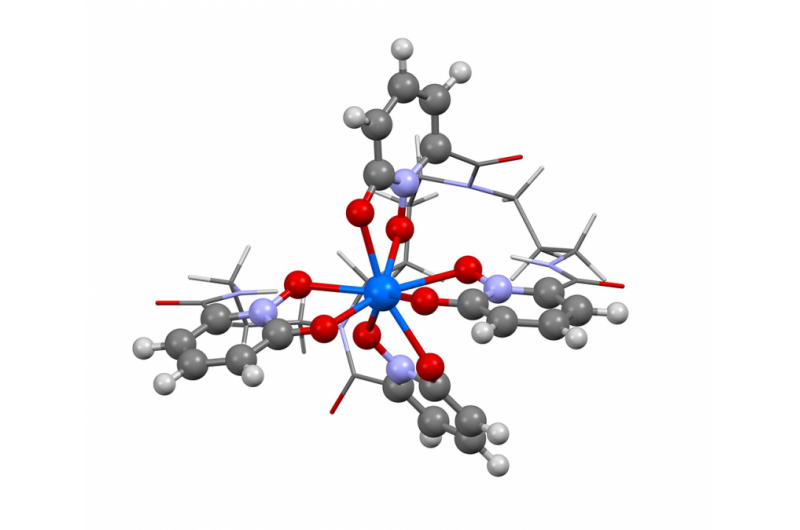
Denna bild visar strukturen av berkelium i oxidationstillstånd +IV. Forskare använde den nya Berkeley Lab-algoritmen för att beräkna absorptionsspektrat och bekräfta vad flera experimentella resultat har antytt - att grundämnet berkelium bryter formen med sina tunga element-kamrater genom att ta en extra positiv laddning när det binds till en syntetisk organisk molekyl. Denna egenskap kan hjälpa forskare att utveckla bättre metoder för att hantera och rena kärnmaterial. Kredit:Bert de Jong, Berkeley Lab
Objekt som lyser i mörkret verkar magiska när du är liten – de kan lysa upp ett mörkt rum utan behov av elektricitet, batterier eller en glödlampa. Sen någon gång lär du dig vetenskapen bakom detta fenomen. Kemiska föreningar som kallas kromoforer får energi, eller upphetsad, när de absorberar synligt ljus. När de återgår till sitt normala tillstånd, den lagrade energin frigörs som ljus, som vi uppfattar som en glöd. Inom materialvetenskap, forskare förlitar sig på ett liknande fenomen för att studera strukturerna hos material som så småningom kommer att användas i kemisk katalys, batterier, solenergiapplikationer och mer.
När en molekyl absorberar en foton – den grundläggande ljuspartikeln – befordras elektroner i molekylsystemet från ett lågenergitillstånd (mark) till ett högre energitillstånd (exciterat). Dessa svar ger resonans vid specifika ljusfrekvenser, lämnar "spektrala fingeravtryck" som belyser de atomära och elektroniska strukturerna i systemet som studeras.
I experiment, "spektrala fingeravtryck" eller absorptionsspektrum, mäts med toppmoderna faciliteter som Advanced Light Source (ALS) vid det amerikanska energidepartementets Lawrence Berkeley National Laboratory (Berkeley Lab). I datorsimuleringar, dessa mätningar fångas vanligtvis med en kvantmekanisk metod som kallas Time Dependent Density Functional Theory (TDDFT). Beräkningsmodellerna är avgörande för att hjälpa forskare att få ut det mesta av sina experiment genom att förutsäga och validera resultat.
Men trots dess användbarhet, det finns tillfällen då TDDFT inte kan användas för att beräkna ett systems absorptionsspektrum eftersom det skulle kräva för mycket tid och datorresurser. Det är här som en ny matematisk "genväg" utvecklad av forskare vid Berkeley Labs Computational Research Division (CRD) kommer väl till pass. Deras algoritm påskyndar absorptionsberäkningar med en faktor på fem, så simuleringar som tidigare tog 10 till 15 timmar att beräkna kan nu göras på cirka 2,5 timmar.
En artikel som beskriver denna metod publicerades i Journal of Chemical Theory and Computation (JCTC). Och det nya tillvägagångssättet för att beräkna absorptionsspektrumet kommer att införlivas i en kommande utgåva av den mycket använda NWChem datorkemiprogramsviten senare i år.
Nya algoritmer leder till beräkningsbesparingar
Att studera den kemiska strukturen hos nya molekyler och material, forskare undersöker vanligtvis systemet med en extern stimulans - vanligtvis en laser - och letar sedan efter små elektroniska förändringar. Matematiskt, denna elektroniska förändring kan uttryckas som ett egenvärdesproblem. Genom att lösa detta egenvärdeproblem, forskare kan få en bra uppskattning av absorptionsspektrumet, vilket i sin tur avslöjar resonansfrekvenserna för systemet som studeras. Under tiden, motsvarande egenvektor används för att beräkna hur intensivt systemet reagerade på stimulansen. Detta är i grunden principen bakom TDDFT-metoden, som har implementerats i flera programvarupaket för kvantkemi, inklusive mjukvarupaketet NWChem med öppen källkod.
Även om detta tillvägagångssätt har visat sig vara framgångsrikt, det har begränsningar för stora system. Ju bredare energiomfång av elektroniska svar en forskare försöker fånga in i ett system, ju fler egenvärden och egenvektorer behöver beräknas, vilket också innebär att fler datorresurser krävs. I sista hand, absorptionsspektrumet för ett molekylärt system med mer än 100 atomer blir oöverkomligt dyrt att beräkna med denna metod.
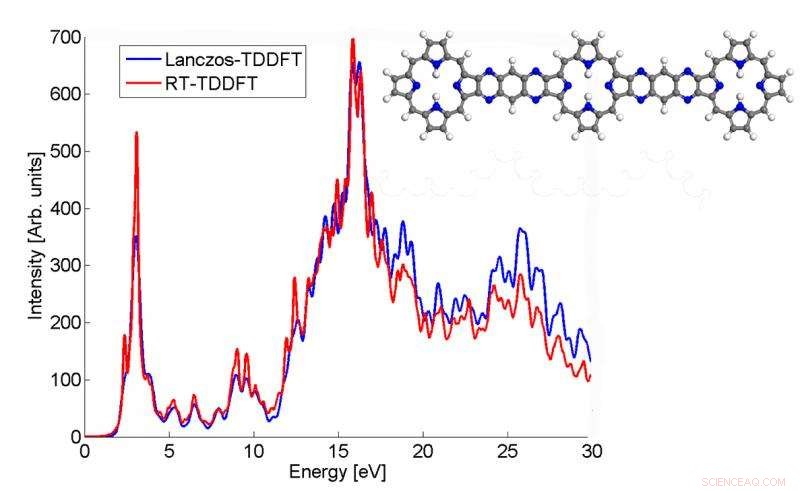
Denna plot visar hur absorptionsspektrumet för en p3b2-molekyl beräknad av Lanczos-algoritmen matchar TDDFT-resultatet i realtid. Kredit:Chao Yang, Berkeley Lab
För att övervinna dessa begränsningar, matematiker i CRD utvecklade en teknik för att beräkna absorptionsspektrumet direkt utan att explicit beräkna matrisens egenvärden.
"Traditionellt, forskare har varit tvungna att beräkna egenvärdena och egenvektorerna för mycket stora matriser för att generera absorptionsspektrum, men vi insåg att du inte behöver beräkna varje enskilt egenvärde för att få en korrekt bild av absorptionsspektrumet, säger Chao Yang, en CRD-matematiker som ledde utvecklingen av det nya tillvägagångssättet.
Genom att omformulera problemet som en matrisfunktionsapproximation, använda sig av en speciell transformation och dra fördel av den underliggande symmetrin med avseende på en icke-euklidisk metrik, Yang och hans kollegor kunde tillämpa Lanczos-algoritmen och en Kernal Polynomial Method (KPM) för att approximera absorptionsspektrumet för flera molekyler. Båda dessa algoritmer kräver relativt lågt minne jämfört med icke-symmetriska alternativ, vilket är nyckeln till de beräkningsmässiga besparingarna.
Eftersom denna metod kräver mindre datorkraft för att uppnå ett resultat, forskare kan också enkelt beräkna absorptionsspektrumet för molekylära system med flera hundra atomer.
"Denna metod är ett viktigt steg framåt eftersom det tillåter oss att modellera absorptionsspektrumet för molekylära system av hundratals atomer till lägre beräkningskostnad." säger Niranjan Govind, en beräkningskemist vid Pacific Northwest National Laboratory som samarbetade med Berkeley Lab-teamet om utvecklingen av metoden i NWChems beräkningskemiprogram.
Nyligen använde Berkeley Lab-forskare den här metoden för att beräkna absorptionsspektrumet och bekräfta vad flera experimentella resultat har antytt - att grundämnet berkelium bryter formen med sina tunga grundämnen genom att ta på sig en extra positiv laddning när det binds till en syntetisk organisk molekyl. Denna egenskap kan hjälpa forskare att utveckla bättre metoder för att hantera och rena kärnmaterial. En artikel som belyser detta resultat dök upp den 10 april i tidskriften Naturkemi .
"De experimentella resultaten antydde detta ovanliga beteende i berkelium, men det fanns inte tillräckligt med experimentella bevis för att säga ja, 100 procent, det här är vad vi ser, " säger studiens medförfattare Wibe Albert de Jong, en CRD-forskare. "För att vara 100 procent säker, vi gjorde stora beräkningssimuleringar och jämförde dem med experimentdata och fastställde att de var, verkligen, ser berkelium i ett ovanligt oxidationstillstånd."
Denna nya algoritm utvecklades genom ett DOE Office of Science-stödt Scientific Discovery through Advanced Computing-projekt (SciDAC) fokuserat på att utveckla programvara och algoritmer för fotokemiska reaktioner. SciDAC-projekt samlar vanligtvis ett tvärvetenskapligt team av forskare för att utveckla nya och nya beräkningsmetoder för att ta itu med några av de mest utmanande vetenskapliga problemen.
"Den tvärvetenskapliga karaktären hos SciDAC är ett mycket effektivt sätt att underlätta banbrytande vetenskap, eftersom varje gruppmedlem ger ett annat perspektiv på problemlösning, " säger Yang. "I denna dynamiska miljö, matematiker, som jag, slå sig samman med domänforskare för att identifiera beräkningsflaskhalsar, sedan använder vi avancerade matematiska tekniker för att hantera och övervinna dessa utmaningar."