MIT-forskare har utvecklat en maskininlärningsmodell som bättre väljer ut molekylkandidater för terapi, samtidigt som det möjliggör automatiserad modifiering av molekylstrukturen för högre styrka. Innovationen har potential att påskynda läkemedelsutvecklingen. Upphovsman:Massachusetts Institute of Technology
Att designa nya molekyler för läkemedel är i första hand en manual, tidskrävande process som är utsatt för fel. Men MIT-forskare har nu tagit ett steg mot att helt automatisera designprocessen, vilket drastiskt kan påskynda saker och ting – och ge bättre resultat.
Läkemedelsupptäckt bygger på blyoptimering. I denna process, kemister väljer en målmolekyl ("bly") med känd potential att bekämpa en specifik sjukdom, justera sedan dess kemiska egenskaper för högre styrka och andra faktorer.
Ofta, kemister använder expertkunskap och utför manuell justering av molekyler, addera och subtrahera funktionella grupper - atomer och bindningar som ansvarar för specifika kemiska reaktioner - en efter en. Även om de använder system som förutsäger optimala kemiska egenskaper, kemister måste fortfarande göra varje modifieringssteg själva. Detta kan ta timmar för varje iteration och kan fortfarande inte producera en giltig läkemedelskandidat.
Forskare från MIT:s Computer Science and Artificial Intelligence Laboratory (CSAIL) och Department of Electrical Engineering and Computer Science (EECS) har utvecklat en modell som bättre väljer blymolekylkandidater baserat på önskade egenskaper. Det modifierar också den molekylära struktur som behövs för att uppnå en högre styrka, samtidigt som man säkerställer att molekylen fortfarande är kemiskt giltig.
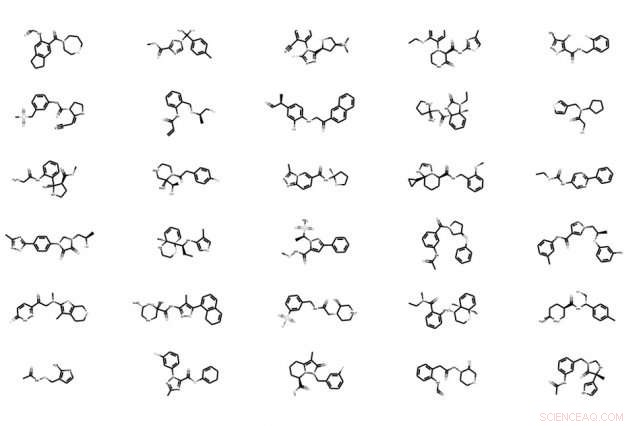
Modellen tar i princip molekylstrukturdata som indata och skapar direkt molekylära grafer – detaljerade representationer av en molekylstruktur, med noder som representerar atomer och kanter som representerar bindningar. Den bryter ner dessa grafer i mindre kluster av giltiga funktionella grupper som den använder som "byggstenar" som hjälper den mer exakt att rekonstruera och bättre modifiera molekyler.
"Motivationen bakom detta var att ersätta den ineffektiva mänskliga modifieringsprocessen för att designa molekyler med automatiserad iteration och säkerställa giltigheten av de molekyler vi genererar, " säger Wengong Jin, en doktorsexamen student i CSAIL och huvudförfattare till ett papper som beskriver modellen som presenteras vid 2018 International Conference on Machine Learning i juli.
Regina Barzilay ansluter sig till Jin på tidningen, Delta Electronics Professor vid CSAIL och EECS och Tommi S. Jaakkola, Thomas Siebel professor i elektroteknik och datavetenskap i CSAIL, EECS, och vid Institutet för data, System, och samhället.
Forskningen utfördes som en del av Machine Learning for Pharmaceutical Discovery and Synthesis Consortium mellan MIT och åtta läkemedelsföretag, meddelade i maj. Konsortiet identifierade lead optimering som en viktig utmaning i läkemedelsupptäckt.
"I dag, det är verkligen ett hantverk, vilket kräver många skickliga kemister för att lyckas, och det är vad vi vill förbättra, " säger Barzilay. "Nästa steg är att ta den här tekniken från akademin till användning på riktiga läkemedelsdesignfall, och visa att det kan hjälpa mänskliga kemister att utföra sitt arbete, vilket kan vara utmanande."
"Att automatisera processen innebär också nya utmaningar för maskininlärning, " säger Jaakkola. "Lär dig relatera, ändra, och generera molekylära grafer driver nya tekniska idéer och metoder. "
Generera molekylära grafer
System som försöker automatisera molekyldesign har dykt upp de senaste åren, men deras problem är giltighet. Dessa system, Jin säger, genererar ofta molekyler som är ogiltiga enligt kemiska regler, och de misslyckas med att producera molekyler med optimala egenskaper. Detta gör i huvudsak full automatisering av molekyldesign omöjlig.
Dessa system körs på linjära notationer av molekyler, kallas "förenklade molekylära ingångssystem för linjeingång, " eller LEDER, där långa bokstäver, tal, och symboler representerar individuella atomer eller bindningar som kan tolkas av datorprogramvara. När systemet modifierar en blymolekyl, den expanderar sin strängrepresentation symbol för symbol - atom för atom, och bind för bindning - tills den genererar en slutlig SMILES-sträng med högre styrka av en önskad egenskap. I slutet, systemet kan producera en slutlig SMILES-sträng som verkar giltig under SMILES-grammatik, men är faktiskt ogiltigt.
Forskarna löser detta problem genom att bygga en modell som körs direkt på molekylgrafer, istället för SMILES-strängar, som kan modifieras mer effektivt och exakt.
Modellen drivs av en anpassad variationsautokodare – ett neuralt nätverk som "kodar" en ingående molekyl till en vektor, som i grunden är ett lagringsutrymme för molekylens strukturella data, och sedan "avkodar" den vektorn till en graf som matchar ingångsmolekylen.
I kodningsfasen, modellen bryter ner varje molekylär graf i kluster, eller "undergrafer, " som var och en representerar en specifik byggsten. Sådana kluster konstrueras automatiskt av ett vanligt maskininlärningskoncept, kallas trädnedbrytning, där en komplex graf mappas till en trädstruktur av kluster - "som ger en ställning av den ursprungliga grafen, " säger Jin.
Både ställningens trädstruktur och molekylära grafstruktur är kodade i sina egna vektorer, där molekyler grupperas genom likhet. Detta gör det lättare att hitta och modifiera molekyler.
Vid avkodningsfasen, modellen rekonstruerar den molekylära grafen på ett "grovt-till-fint" sätt - gradvis ökande upplösning av en lågupplöst bild för att skapa en mer förfinad version. Det genererar först den trädstrukturerade ställningen, och monterar sedan de tillhörande klustren (noder i trädet) tillsammans till en koherent molekylgraf. Detta säkerställer att den rekonstruerade molekylära grafen är en exakt replikering av den ursprungliga strukturen.
För leadoptimering, modellen kan sedan modifiera blymolekyler baserat på en önskad egenskap. Det gör det med hjälp av en förutsägelsealgoritm som ger varje molekyl poäng med ett potensvärde för den egenskapen. I tidningen, till exempel, forskarna sökte molekyler med en kombination av två egenskaper – hög löslighet och syntetisk tillgänglighet.
Med tanke på en önskad egenskap, modellen optimerar en ledande molekyl genom att använda prediktionsalgoritmen för att modifiera dess vektor—och, därför, struktur – genom att redigera molekylens funktionella grupper för att uppnå en högre potenspoäng. Det upprepar detta steg i flera iterationer, tills den hittar den högsta förutsagda potenspoängen. Sedan, modellen avkodar slutligen en ny molekyl från den uppdaterade vektorn, med modifierad struktur, genom att sammanställa alla motsvarande kluster.
Giltig och mer potent
Forskarna tränade sin modell på 250, 000 molekylära grafer från ZINC-databasen, en samling 3D-molekylära strukturer tillgängliga för allmänt bruk. De testade modellen på uppgifter för att generera giltiga molekyler, hitta de bästa blymolekylerna, och designa nya molekyler med ökande styrkor.
I det första testet, forskarnas modell genererade 100 procent kemiskt giltiga molekyler från en provfördelning, jämfört med SMILES -modeller som genererade 43 procent giltiga molekyler från samma distribution.
Det andra testet innebar två uppgifter. Först, modellen sökte igenom hela samlingen av molekyler för att hitta den bästa blymolekylen för de önskade egenskaperna – löslighet och syntetisk tillgänglighet. I den uppgiften modellen hittade en blymolekyl med 30 procent högre styrka än traditionella system. Den andra uppgiften innebar att modifiera 800 molekyler för högre styrka, men är strukturellt lik blymolekylen. Genom att göra så, modellen skapade nya molekyler, liknar ledningens struktur, i genomsnitt en förbättring av styrkan med mer än 80 procent.
Forskarna syftar sedan till att testa modellen på fler egenskaper, bortom löslighet, som är mer terapeutiskt relevanta. Den där, dock, kräver mer data. "Läkemedelsföretag är mer intresserade av fastigheter som kämpar mot biologiska mål, men de har mindre data om dem. En utmaning är att utveckla en modell som kan fungera med en begränsad mängd träningsdata, " säger Jin.
Den här historien återpubliceras med tillstånd av MIT News (web.mit.edu/newsoffice/), en populär webbplats som täcker nyheter om MIT-forskning, innovation och undervisning.