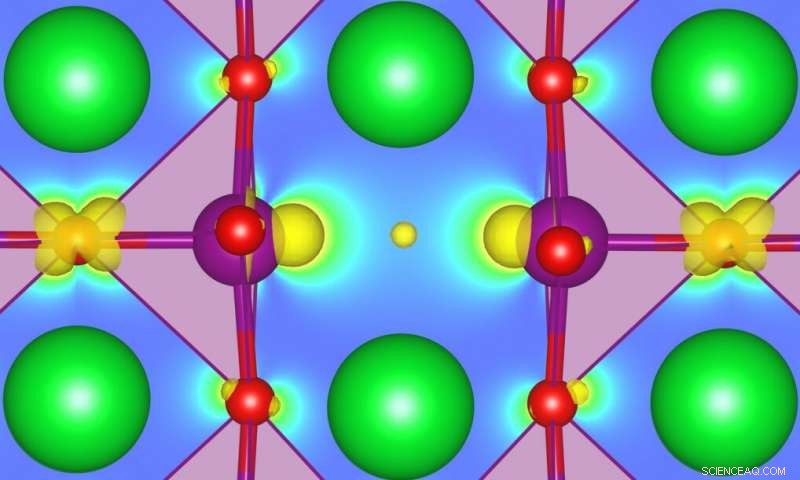
Förstå hur defekter kan påverka marktillståndsegenskaper, främja fasövergångar, eller möjliggöra helt nya funktionaliteter i vissa starkt korrelerade oxider har blivit ett ämne av stort intresse inom området design och upptäckt av nya funktionella material. SrMnO3 (SMO) är ett särskilt intressant exempel, men bättre karakterisering behövs. MARVEL-forskare har nu utvecklat en metod som kan leda till mer exakta förutsägelser av energin hos defekter som är associerade med in-gap-tillstånd i halvledare eller isolatorer. Kredit:Ulrich Aschauer
Förstå hur defekter kan påverka marktillståndsegenskaper, främja fasövergångar, eller möjliggöra helt nya funktionaliteter i vissa starkt korrelerade oxider har blivit ett ämne av stort intresse inom området design och upptäckt av nya funktionella material. SrMnO 3 (SMO) är ett särskilt intressant exempel, men bättre karakterisering behövs. MARVEL-forskare har nu utvecklat en metod som kan leda till mer exakta förutsägelser av energin hos defekter associerade med in-gap-tillstånd i halvledare eller isolatorer.
Några perovskitoxider, till exempel, har visat ett brett spektrum av tekniskt relevanta funktionella egenskaper som ferroelektricitet och magnetism som kan ställas in via stam. Anstränga, dock, kopplar också ihop med defektkemin för att bestämma materialets egenskaper.
SrMnO 3 (SMO) är ett särskilt intressant exempel för att undersöka funktionaliteten som härrör från ett komplext samspel av stam, magnetisk ordning, polära förvrängningar, och syrevakanser som är allestädes närvarande defekter i dessa material. Särskilt, teorin har förutsagt att SMO -tunna filmer ska övergå från antiferromagnetiska till ferromagnetiska med ökande syrebrist, vilket stöds av nyare experimentella studier.
Dessa tidigare förutsägelser var dock baserade på densitetsfunktionella teorin (DFT) beräkningar som inkluderade en korrigering U baserad på de elektroniska och magnetiska egenskaperna hos stökiometriska manganiter. Även om införandet av U - avsett att korrigera självinteraktion av elektroner i komplexa oxider - är nödvändigt i sådana material, det specifika valet av U baserat på stökiometriska materialegenskaper kan leda till potentiella brister i beskrivningen av defekt SMO—manganjoner runt defekten har en annan koordinationsmiljö.
Beroende på defekt laddningstillstånd, ett extra problem är relaterat till beskrivningen av flera oxidationstillstånd som finns i defekt SMO. Bildandet av syrevakanser kompenseras i allmänhet genom en minskning av oxidationstillståndet (OS) för manganjoner intill vakansen, som alltså inte kan beskrivas korrekt av samma U.
Det är därför Chiara Ricca och kollegor vid universitetet i Bern beslutade att det var avgörande att ta hänsyn till lokala strukturella och kemiska effekter för varje övergångsmetallplats i oxiden när man strävar efter en korrekt beskrivning av defekt SMO. I samarbete med ett team på Nicola Marzaris THEOS -lab, som nyligen utvecklade en densitetsfunktionella störningsteori (DFPT)-baserad metod för att beräkna U-parametrar, de använde självständiga platsberoende U-värden beräknade från första principer för att studera defektkemin och magnetiska egenskaper hos SMO-bulk och spända tunna filmer.
"Detta extremt nära samarbete mellan de två grupperna, den ena fokuserar på metodutveckling och den andra på tillämpningar i defekta oxidmaterial, utlöstes av att förena dessa olika forskningsfokus under MARVEL-paraplyet", sa Ulrich Aschauer vid universitetet i Bern, en av de två PI:er som är involverade i arbetet.
Resultaten visar att detta självständiga U förbättrar strukturen av stökiometrisk SrMnO 3 med hänsyn till andra metoder, inklusive en som använder ett empiriskt U. För defekta system, U ändras som en funktion av avståndet mellan övergångsmetallplatsen från defekten, dess oxidationstillstånd, dess koordinationsnummer, och materialets magnetiska fas. Med hänsyn till detta beroende, i tur och ordning, påverkar de beräknade defektbildningsenergierna och de förutsagda töjnings- och/eller defektinducerade magnetiska fasövergångarna, speciellt när ockuperade lokaliserade tillstånd uppträder i materialets bandgap vid skapandet av defekter.
"Vi tror att detta tillvägagångssätt kan leda till mer exakta förutsägelser av energin hos defekter associerade med in-gap-tillstånd i halvledare eller isolatorer både jämfört med standard DFT och möjligen hybridfunktioner till en beräkningskostnad som är betydligt lägre än för den senare, "Ricca sa." Detta är tack vare en korrekt beskrivning av de strukturella och lokala kemiska effekter som orsakas av defekterna. "