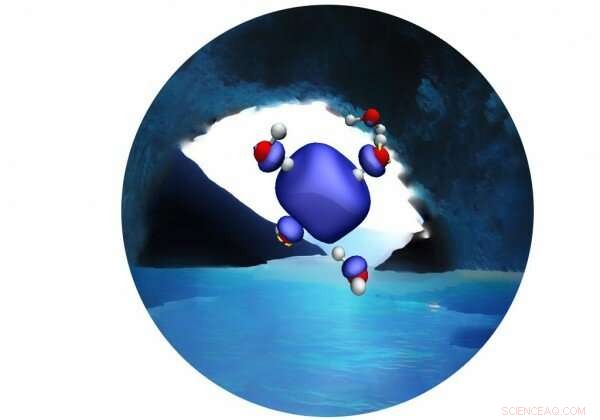
En ny MD-simulering ger avgörande bevis till förmån för en ihållande tetraedrisk hålighet som består av fyra vattenmolekyler. Kredit:Vladimir Rybkin
Extra elektroner lösta i flytande vatten, kända som hydratiserade elektroner, rapporterades första gången för 50 år sedan. Dock, deras struktur är fortfarande inte väl förstådd. MARVEL-forskare vid universitetet i Zürich, ETH och Swiss National Supercomputing Center CSCS har nu tagit ett steg mot att lösa mysteriet. Deras papper, "Dynamiken för den bulkhydrerade elektronen från många kroppsvågsfunktionsteori, " har publicerats i Angewandte Chemie .
Den e - aq arten är svår att observera direkt eftersom den är kortlivad och inte kan separeras eller koncentreras. Detta utesluter användning av direkta strukturella tillvägagångssätt, diffraktion eller NMR-spektroskopi för att utforska dess struktur. Även om vissa egenskaper inklusive spektra i UV- och IR-regioner och bindningsenergin har observerats direkt, den allmänna bristen på direkta experimentella mätningar av strukturen hos den hydratiserade elektronen kräver teori.
Tillförlitlig modellering av den hydratiserade elektronen är minst lika utmanande som den experimentella metoden, och begränsningarna av beräkningsmetoder som hittills tillämpats har lett till avsevärd teoretisk osäkerhet. Forskare har inte, till exempel, kunnat komma överens om huruvida den hydratiserade elektronen upptar en kavitet eller inte. Även om de flesta teoretiska studier tyder på att det gör det, modeller utan kavitet har också visat sig vara korrekta. En annan diskussionspunkt är kopplad till de urskiljbara yt- och bulkstrukturerna hos den hydratiserade elektronen.
I tidningen, forskarna Vladimir Rybkin och Jan Wilhelm vid universitetet i Zürich och Joost VadeVondele vid ETH Zürich och CSCS använde den första molekylära dynamiksimuleringen av den bulkhydratiserade elektronen baserad på korrelerad vågfunktionsteori för att ge avgörande bevis till förmån för en ihållande tetraedrisk kavitet som består av fyra vattenmolekyler. De visade också att det inte finns några stabila icke-kavitetsstrukturer i den bulkhydratiserade elektronen.
Forskarna kom fram till sin modell genom att noggrant överväga vilka egenskaper det mest exakta tillvägagångssättet måste ha. De ville att det skulle baseras på molekylär dynamik för att fånga bildningen och dynamiska transformationer av kaviteten. De behövde en mångakroppskorrelerad elektronisk strukturnivå för att undvika delokaliseringsfel och för att möjliggöra de korrelationseffekter som har befunnits vara avgörande för att förutsäga lösningen av elektronen exakt utan empiriska parametrar. De ville att simuleringen skulle utföras i bulken under periodiska randförhållanden för att undvika bildning av ytstrukturen och, till sist, metoden bör ge en korrekt beskrivning av flytande vatten.
MD-simuleringen uppfyller alla dessa krav. Den representerar den första dynamiska simuleringen av en komplex kemisk art i den kondenserade fasen på den korrelerade vågfunktionsnivån i teorin. Detta var det första kritiska steget. Den andra genomförde faktiskt det. Sådana beräkningar har varit tekniskt omöjliga tills de senaste framstegen – inklusive de som gjorts i deras egna grupper – har möjliggjort massivt parallella teoriberäkningar av många kroppar i den förtätade fasen på toppmoderna superdatorer som de som CSCS. Det tog dem ändå ungefär 1 miljon nodtimmar på superdatorn Piz Daint, Europas snabbaste.
Modellen visade att en kavitet bildas inom 250 femtosekunder efter att överskottselektronen tillförts det ostörda flytande vattnet. Kritiskt, simuleringen misslyckades med att hitta några bevis för icke-kavitetsstrukturer hos den bulkhydratiserade elektronen i varken stabila eller metastabila tillstånd. Detta ger mycket starkare teoretiska bevis för kavitetsmodellen.