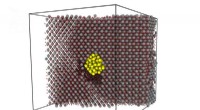
Använda superdatorer, forskare har precis börjat designa proteiner som självbildar sig för att kombinera och likna livgivande molekyler som hemoglobin. Kredit:Taylor et al.
Röda blodkroppar är fantastiska. De plockar upp syre från våra lungor och bär det över hela kroppen för att hålla oss vid liv. Hemoglobinmolekylen i röda blodkroppar transporterar syre genom att ändra dess form på ett allt-eller-inget-sätt. Fyra kopior av samma protein i hemoglobin öppnar och stänger som blomblad, strukturellt kopplade för att svara på varandra. Använda superdatorer, forskare har precis börjat designa proteiner som självbildar sig för att kombinera och likna livgivande molekyler som hemoglobin. Forskarna säger att deras metoder kan tillämpas på användbara tekniker som läkemedelsmålning, artificiell energiskörd, "smart" avkänning och byggmaterial, och mer.
Ett vetenskapsteam gjorde detta arbete genom att överladda proteiner, vilket betyder att de ändrade underenheterna av proteiner, aminosyrorna, för att ge proteinerna en artificiellt hög positiv eller negativ laddning. Använda proteiner som härrör från maneter, forskarna kunde sätta ihop en komplex struktur med sexton proteiner som består av två staplade oktamerer genom enbart överladdning, fynd som rapporterades i januari 2019 i tidskriften Naturkemi .
Teamet använde sedan superdatorsimuleringar för att validera och informera dessa experimentella resultat. Superdatortilldelningar på Stampede2 vid Texas Advanced Computing Center (TACC) och Comet vid San Diego Supercomputer Center (SDSC) tilldelades forskarna genom XSEDE, Extreme Science and Engineering Discovery Environment finansierad av National Science Foundation (NSF).
"Vi fann att genom att ta proteiner som normalt inte interagerar med varandra, vi kan göra kopior som antingen är mycket positivt eller mycket negativt laddade, " sa studiens medförfattare Anna Simon, en postdoktor vid Ellington Lab vid UT Austin. " Genom att kombinera de mycket positivt och negativt laddade kopiorna, vi kan få proteinerna att samlas till mycket specifika strukturerade sammansättningar, " sa Simon. Forskarna kallar sin strategi "överladdad proteinsammansättning, ' där de driver definierade proteininteraktioner genom att kombinera konstruerade överladdade varianter.
"Vi utnyttjade en mycket välkänd och grundläggande princip från naturen, att motsatta laddningar lockar, " tillade studiens medförfattare Jens Glaser. Glaser är biträdande forskare i Glotzer Group, Institutionen för kemiteknik vid University of Michigan. "Anna Simons grupp fann att när de blandar dessa laddade varianter av grönt fluorescerande protein, de får mycket ordnade strukturer. Det var en riktig överraskning, " sa Glaser.
Den staplade oktamerstrukturen ser ut som en flätad ring. Den består av 16 proteiner - två sammanflätade ringar av åtta som interagerar i mycket specifika, diskreta plåster. "Anledningen till att det är så svårt att konstruera proteiner som interagerar syntetiskt är att man gör dessa interagerande fläckar och får dem alla i rätt linje så att de tillåter proteinerna att samlas till större, vanliga strukturer är riktigt svårt, " förklarade Simon. De kom runt problemet genom att lägga till många positiva och negativa laddningar till ingenjörsvarianter av grönt fluorescerande protein (GFP), ett välstuderat "labmus"-protein som härrör från maneten Aequorea victoria.
Det positivt laddade proteinet, som de kallade cerulean fluorescent protein (Ceru) +32, hade ytterligare möjligheter att interagera med det negativt laddade proteinet GFP -17. "Genom att ge dessa proteiner alla dessa möjligheter, dessa olika platser där de potentiellt kan interagera, de kunde välja de rätta, " sa Simon. "Det fanns vissa mönster och interaktioner som fanns där, tillgängliga, och energiskt gynnade, som vi inte nödvändigtvis förutspådde i förväg som skulle tillåta dem att monteras till dessa specifika former."
För att få de konstruerade laddade fluorescerande proteinerna, Simon och medförfattarna Arti Pothukuchy, Jimmy Gollihar, och Barrett Morrow kodade deras gener, inklusive en kemisk etikett som används för rening på bärbara bitar av DNA som kallas plasmider i E. coli, skördade sedan det märkta proteinet som E. coli odlade. Forskarna blandade ihop proteinerna. De trodde först att proteinerna bara kunde interagera för att bilda stora, oregelbundet strukturerade klumpar. "Men då, det vi hela tiden såg var så konstigt, rolig topp runt 12 nanometer, som var mycket mindre än en stor proteinklump, men betydligt större än det enskilda proteinet, sa Simon.
De mätte storleken på partiklarna som bildades med hjälp av ett Zetasizer-instrument vid Texas Materials Institute of UT Austin, och verifierade att partiklarna innehöll både cerulean- och GFP-proteiner Förster Resonance Energy Transfer (FRET), som mäter energiöverföringen mellan olika färgade fluorescerande proteiner producerar fluorescens som svar på olika energier av ljus för att se om de är nära varandra. Negativ fläckelektronmikroskopi identifierade partiklarnas specifika struktur, dirigerad av gruppen av David Taylor, biträdande professor i molekylär biovetenskap vid UT Austin. Den visade att 12 nm-partikeln bestod av en staplad oktamer sammansatt av sexton proteiner. "Vi upptäckte att de var dessa vackert formade blomliknande strukturer, " Sa Simon. Medförfattaren Yi Zhou från Taylors grupp av UT Austin ökade upplösningen ytterligare med hjälp av kryoelektronmikroskopi för att avslöja detaljer på atomnivå av den staplade oktameren.

XSEDE gav forskare tillgång till Comet-superdatorn vid San Diego Supercomputer Center (vänster) och Stampede2-superdatorn vid Texas Advanced Computing Center (höger). Kredit:SDSC, TACC
Beräkningsmodellering förfinade mätningarna av hur proteinerna var ordnade till en tydlig bild av det vackra, blomliknande struktur, enligt Jens Glaser. "Vi var tvungna att ta fram en modell som var tillräckligt komplex för att beskriva fysiken hos de laddade gröna fluorescerande proteinerna och presentera alla relevanta atomistiska detaljer, men var tillräckligt effektiv för att tillåta oss att simulera detta på en realistisk tidsskala. Med en helt atomistisk modell, det skulle ha tagit oss över ett år att få ut en enda simulering från datorn, hur snabb datorn än var, " sa Glaser.
De förenklade modellen genom att minska upplösningen utan att offra viktiga detaljer om interaktionerna mellan proteiner. "Det är därför vi använde en modell där formen på proteinet representeras exakt av en molekylär yta, precis som den som mäts från proteinets kristallografiska struktur, " tillade Glaser.
"Det som verkligen hjälpte oss att vända detta och förbättra vad vi kunde få ut av våra simuleringar var cryo-EM-data, " sa Vyas Ramasubramani, en doktorand i kemiteknik vid University of Michigan. "Det var det som verkligen hjälpte oss att hitta den optimala konfigurationen att lägga in i dessa simuleringar, som sedan hjälpte oss att validera stabilitetsargumenten som vi gjorde, och förhoppningsvis framöver göra förutsägelser om hur vi kan destabilisera eller modifiera denna struktur, " sa Ramasubramani.
Forskarna krävde massor av beräkningskraft för att göra beräkningarna i den skala de ville ha.
"Vi använde XSEDE för att i princip ta dessa enorma system, där du har många olika delar som interagerar med varandra, och beräkna allt detta på en gång så att när du börjar flytta ditt system framåt genom en viss tid, du kan få en uppfattning om hur det kommer att utvecklas på lite verkliga tidsskalor, " sa Ramasubramani. "Om du försökte göra samma typ av simulering som vi gjorde på en bärbar dator, det skulle ha tagit månader om inte år att verkligen närma sig att förstå huruvida någon form av struktur skulle vara stabil eller inte. För oss, att inte kunna använda XSEDE, där du kan använda i huvudsak 48 kärnor, 48 beräkningsenheter på en gång för att göra dessa beräkningar mycket parallella, vi skulle ha gjort det här mycket långsammare."
Superdatorn Stampede2 vid TACC innehåller 4, 200 Intel Knights Landing och 1, 736 Intel Skylake X beräkningsnoder. Varje Skylake-nod har 48 kärnor, grundenheten i en datorprocessor. "Skylake-noderna i superdatorn Stampede2 var avgörande för att uppnå den prestanda som var nödvändig för att beräkna dessa elektrostatiska interaktioner som verkar mellan de motsatt laddade proteinerna på ett effektivt sätt, ", sa Glaser. "Tillgängligheten för superdatorn Stampede2 var vid precis rätt tidpunkt för oss att utföra dessa simuleringar."
Initialt, forskarteamet testade sina simuleringar på Comet-systemet vid SDSC. "När vi först tänkte ut vilken typ av modell vi skulle använda och om den här förenklade modellen skulle ge oss rimliga resultat, Comet var ett bra ställe att prova dessa simuleringar, "Sa Ramasubramani. "Comet var en stor testbädd för vad vi gjorde."
Ser man på den större vetenskapliga bilden, the scientists hope that this work advances understanding of why so many proteins in nature will oligomerize, or join together to form more complex and interesting structures.
"We showed that there doesn't need to be a very specific, pre-distinguished set of plans and interactions for these structures to form, " Simon said. "This is important because it means that maybe, and quite likely we can take other sets of molecules that we want to make oligomerize and generate both positively charged and negatively charged variants, combine them, and have specifically ordered structures for them."
Natural biomaterials like bone, feathers, and shells can be tough yet lightweight. "We think supercharged protein assembly is an easier way to develop the kind of materials that have exciting synthetic properties without having to spend so much time or having to know exactly how they're going to come together beforehand, " Simon said. "We think that will accelerate the ability to engineer synthetic materials and for discovery and exploration of these nanostructured protein materials."
Studien, "Supercharging Enables Organized Assembly of Synthetic Biomolecules, " was published in the journal Naturkemi in January of 2019.