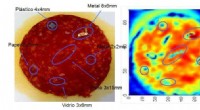
Forskare utvecklade en maskininlärningsalgoritm för att förutsäga 3D-molekylär kristalldensitet från kemiska 2D-strukturer. Kredit:Lawrence Livermore National Laboratory
Ett långvarigt mål av kemister inom många branscher, inklusive energi, läkemedel, energi, livsmedelstillsatser och organiska halvledare, är att föreställa sig den kemiska strukturen hos en ny molekyl och kunna förutsäga hur den kommer att fungera för en önskad tillämpning. I praktiken, denna vision är svår, kräver ofta omfattande laboratoriearbete för att syntetisera, isolera, rena och karakterisera nydesignade molekyler för att få önskad information.
Nyligen, ett team av Lawrence Livermore National Laboratory (LLNL) material och datavetare har förverkligat denna vision för energiska molekyler genom att skapa modeller för maskininlärning (ML) som kan förutsäga molekylers kristallina egenskaper enbart utifrån deras kemiska strukturer, såsom molekylär densitet. Att förutsäga kristallstrukturdeskriptorer (snarare än hela kristallstrukturen) erbjuder en effektiv metod för att härleda ett material egenskaper, vilket påskyndar materialdesign och upptäckt. Forskningen visas i Journal of Chemical Information and Modeling .
"En av teamets mest framträdande ML-modeller är kapabel att förutsäga den kristallina densiteten av energiska och energiliknande molekyler med en hög grad av noggrannhet jämfört med tidigare ML-baserade metoder, sade Phan Nguyen, LLNL tillämpad matematiker och co-första författare av uppsatsen.
"Även jämfört med densitetsfunktionella teorin (DFT), en beräkningsmässigt dyr och fysikinformerad metod för förutsägelse av kristallstruktur och kristallina egenskaper, ML-modellen har konkurrenskraftig noggrannhet samtidigt som den kräver en bråkdel av beräkningstiden, sa Donald Loveland, LLNL datavetare och medförfattare.
Medlemmar av LLNL:s High Explosive Application Facility (HEAF) har redan börjat dra nytta av modellens webbgränssnitt, med ett mål att upptäcka nya okänsliga energimaterial. Genom att helt enkelt mata in molekylernas 2D-kemiska struktur, HEAF-kemister har snabbt kunnat bestämma den förutsagda kristallina densiteten för dessa molekyler, vilket är nära korrelerat med potentiella energiers prestationsmått.
"Vi är glada över att se resultaten av vårt arbete tillämpas på viktiga uppdrag i labbet. Detta arbete kommer säkerligen att hjälpa till att påskynda upptäckten och optimeringen av nya material framåt, sa Yong Han, LLNL materialforskare och huvudutredare för projektet.
Uppföljningsinsatser inom materialvetenskapsavdelningen har använt ML-modellen i kombination med en generativ modell för att snabbt och effektivt söka i stora kemiska utrymmen efter högdensitetskandidater.
"Båda ansträngningarna tänjer på gränserna för materialupptäckt och underlättas genom det nya paradigmet att slå samman materialvetenskap och maskininlärning, sa Anna Hiszpanski, LLNL materialforskare och medförfattare till artikeln.
Teamet fortsätter att söka efter nya egenskaper av intresse för labbet med visionen att tillhandahålla en svit av prediktiva modeller för materialforskare att använda i sin forskning.