Det finns få problem nu som AI och maskininlärning inte kan hjälpa till att övervinna. Forskare från Yokohama National University använder denna moderna fördel för att lösa vad konventionella metoder inte kan.
Det finns många regler att komma ihåg när det kommer till växelverkan mellan kolhaltiga (eller organiska) molekyler:placeringen av grupper på molekylen som interagerar med dess omgivning, molekylens storlek, form och position, och molekylen med vilken det interagerar. Resultatet av en given reaktion kan vara väldigt olika beroende på dessa faktorer och många fler, och att förutsäga dessa utfall har visat sig vara en ganska stor utmaning inom det kemiska området. Att kontrollera resultatet är en mycket nödvändig komponent i kemisk syntes, men förutsägelser räcker inte alltid.
Lyckligtvis kan maskininlärning och artificiell intelligens (AI) återigen hjälpa till att driva framsteg framåt genom att förutsäga hastigheten eller selektiviteten för en given reaktion. Därför kan denna teknik vara användbar för att förutsäga vilken produkt som kan förväntas.
Forskarna har publicerat sina resultat i Journal of Chemical Information and Modeling .
I organisk kemi är varje detalj viktig. Två vanliga områden som kan påverka hur en molekyl samverkar med andra molekyler är steriska och orbitaler. Steriska hänvisar till arrangemanget av molekyler och steriska effekter kan bestämma formen och reaktiviteten hos molekylen. Detta kan bero på storleken eller laddningen av molekylen eller den enskilda atomen. Orbitaler är ett sätt att förklara den mest sannolika placeringen av elektronerna som i sin tur kan interagera med andra molekyler eller atomer för att orsaka reaktioner.
Dessa faktorer kan drastiskt förändras där en nukleofil, eller en elektrondonerande reaktant, kan fästa till mottagarmolekylen. Detta är känt som "selektivitet", och beroende på var molekylen fäster kan resultaten bilda olika produkter eller utbyten av den önskade produkten. Forskare använder AI och maskininlärning samt den nuvarande kunskapen om kemiska reaktioner för att bättre förklara dessa aspekter av molekylär selektivitet.
"För att avgöra vilken information som kan användas som väsentlig kemisk information som ska ges till AI, är det nödvändigt att kombinera kemisk kunskap med kunskap om AI och maskininlärning", säger motsvarande författare Hiroaki Gotoh, docent vid tekniska fakulteten, Yokohama National University.
Först måste datorn matas med information som man kan lära sig från. Information från litteratur inom beräkningskemi och information från tidigare studier användes för att påbörja undervisningsprocessen för AI. Efter en del manuell inmatning av data för de specifika molekylerna som används och inställning av optimala parametrar, kördes dataanalyser baserat på de förutspådda resultaten av testdatauppsättningen. Dessa analyser gör det möjligt för AI att lära sig och förutsäga framtida selektiviteter baserat på redan känd information.
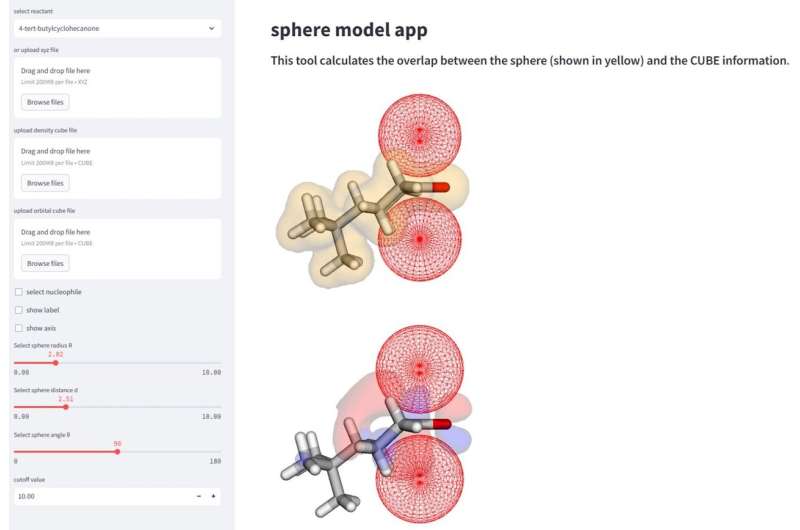
"Denna metod möjliggör en mer omfattande analys och tolkning av reaktionsmekanismer via beräkning av parametrarna för de sfäriska utrymmena som efterliknar närmar sig nukleofiler", säger Daimon Sakaguchi, första författare till studien vid institutionen för kemi och biovetenskap, Yokohama National University.
Studien förklarade framgångsrikt 323-reaktionsselektiviteten hos åtta nukleofiler, baserat på vilken "ansikte" av molekylen som skulle ge den mest önskvärda mängden produkt. Selektiviteten förändras baserat på molekylens steriska egenskaper förutom dess orbitala faktorer. Forskare fann att för vissa molekyler är orbitalfaktorn viktigare för att bestämma ansiktsselektivitet, och andra är mer beroende av molekylens steriska egenskaper när den interagerar med sin nukleofil.
Kombinationen av prediktiv teknik och maskininlärning med etablerad kunskap om kemi kan ge bättre resultat från den kemiska reaktionen och hjälpa kemister att syntetisera naturliga produkter och farmaceutiska kemikalier på ett mer strömlinjeformat sätt.
Genom att effektivisera denna process med användning av maskininlärning och artificiell intelligens kan mer experimenterande ske. Helst hoppas forskarna att samarbeta med experimentella kemister för att utforma reaktioner som kommer att fortsätta med utvecklingen av mer prediktiv teknologi för kemiska reaktioner.
Mer information: Daimon Sakaguchi et al, Användning av tredimensionell information för att förutsäga och tolka ansiktsselektiviteten hos nukleofila tillägg till cykliska ketoner, Journal of Chemical Information and Modeling (2024). DOI:10.1021/acs.jcim.4c00101
Journalinformation: Journal of Chemical Information and Modeling
Tillhandahålls av Yokohama National University