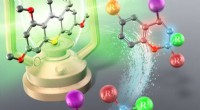
Ett nytt automatiserat arbetsflöde utvecklat av forskare vid Lawrence Berkeley National Laboratory (Berkeley Lab) har potential att tillåta forskare att analysera produkterna från sina reaktionsexperiment i realtid, en nyckelfunktion som behövs för framtida automatiserade kemiska processer.
Det utvecklade arbetsflödet – som tillämpar statistisk analys för att bearbeta data från kärnmagnetisk resonans (NMR) spektroskopi – kan hjälpa till att påskynda upptäckten av nya läkemedel och påskynda utvecklingen av nya kemiska reaktioner.
Forskarna från Berkeley Lab som utvecklade den banbrytande tekniken säger att arbetsflödet snabbt kan identifiera den molekylära strukturen hos produkter som bildas av kemiska reaktioner som aldrig har studerats tidigare. De rapporterade nyligen sina fynd i Journal of Chemical Information and Modeling .
Förutom läkemedelsupptäckt och utveckling av kemiska reaktioner kan arbetsflödet också hjälpa forskare som utvecklar nya katalysatorer. Katalysatorer är ämnen som underlättar en kemisk reaktion vid produktion av användbara nya produkter som förnybara bränslen eller biologiskt nedbrytbar plast.
"Det som upphetsar människor mest med den här tekniken är dess potential för reaktionsanalys i realtid, som är en integrerad del av automatiserad kemi", säger första författaren Maxwell C. Venetos, en före detta forskare vid Berkeley Labs Materials Sciences Division och tidigare doktorandstudent. forskare i materialvetenskap vid UC Berkeley. Han avslutade sina doktorandstudier förra året.
"Vårt arbetsflöde låter dig verkligen börja sträva efter det okända. Du är inte längre begränsad av saker som du redan vet svaret på."
Det nya arbetsflödet kan också identifiera isomerer, som är molekyler med samma kemiska formel men olika atomarrangemang. Detta skulle kunna påskynda syntetiska kemiska processer i till exempel läkemedelsforskning.
"Det här arbetsflödet är det första i sitt slag där användare kan skapa sitt eget bibliotek och anpassa det till kvaliteten på det biblioteket utan att förlita sig på en extern databas," sa Venetos.
Inom läkemedelsindustrin använder läkemedelsutvecklare för närvarande maskinlärande algoritmer för att virtuellt screena hundratals kemiska föreningar för att identifiera potentiella nya läkemedelskandidater som är mer sannolikt att vara effektiva mot specifika cancerformer och andra sjukdomar. Dessa screeningmetoder går igenom onlinebibliotek eller databaser med kända föreningar (eller reaktionsprodukter) och matchar dem med troliga läkemedels-"mål" i cellväggarna.
Men om en läkemedelsforskare experimenterar med molekyler så nya att deras kemiska strukturer ännu inte finns i en databas, måste de vanligtvis tillbringa dagar i labbet för att reda ut blandningens molekylära sammansättning. Först genom att köra reaktionsprodukterna genom en reningsmaskin och sedan använda ett av de mest användbara karaktäriseringsverktygen i en syntetisk kemists arsenal, en NMR-spektrometer, för att identifiera och mäta molekylerna i blandningen en i taget.
"Men med vårt nya arbetsflöde kan du göra allt detta arbete inom ett par timmar," sa Venetos. Tidsbesparingarna kommer från arbetsflödets förmåga att snabbt och noggrant analysera NMR-spektra för orenade reaktionsblandningar som innehåller flera föreningar, en uppgift som är omöjlig genom konventionella NMR-spektralanalysmetoder.
"Jag är väldigt exalterad över det här arbetet eftersom det tillämpar nya datadrivna metoder på det urgamla problemet med att accelerera syntes och karakterisering", säger seniorförfattaren Kristin Persson, seniorforskare på fakulteten vid Berkeley Labs materialvetenskapsavdelning och professor vid UC Berkeley. av materialvetenskap och ingenjörskonst som också leder materialprojektet.
Förutom att det är mycket snabbare än reningsmetoder på bänkskivor, har det nya arbetsflödet potential att bli lika exakt. NMR-simuleringsexperiment utförda med hjälp av National Energy Research Scientific Computing Center (NERSC) vid Berkeley Lab med stöd från Materials Project visade att det nya arbetsflödet korrekt kan identifiera sammansatta molekyler i reaktionsblandningar som producerar isomerer och även förutsäga de relativa koncentrationerna av dessa föreningar.
För att säkerställa hög statistisk noggrannhet använde forskargruppen en sofistikerad algoritm känd som Hamiltonian Monte Carlo Markov Chain (HMCMC) för att analysera NMR-spektra. De utförde även avancerade teoretiska beräkningar baserade på en metod som kallas densitetsfunktionella teorin.
Venetos designade det automatiserade arbetsflödet som öppen källkod så att användare kan köra det på en vanlig stationär dator. Den bekvämligheten kommer väl till pass för alla från industrin eller den akademiska världen.
Tekniken växte fram från samtal mellan Persson-gruppen och experimentella medarbetare Masha Elkin och Connor Delaney, tidigare postdoktorer i John Hartwig-gruppen vid UC Berkeley. Elkin är nu professor i kemi vid Massachusetts Institute of Technology och Delaney professor i kemi vid University of Texas i Dallas.
"I utvecklingen av kemireaktioner lägger vi ständigt tid på att ta reda på vad en reaktion skapade och i vilket förhållande", säger John Hartwig, senior forskare vid Berkeley Labs avdelning för kemiska vetenskaper och professor i kemi vid UC Berkeley.
"Vissa NMR-spektrometrimetoder är exakta, men om man dechiffrerar innehållet i en rå reaktionsblandning som innehåller ett gäng okända potentiella produkter, är dessa metoder alldeles för långsamma för att ha som en del av ett experimentellt eller automatiserat arbetsflöde med hög genomströmning. Och det är där den här nya förmågan att förutsäga NMR-spektrumet skulle kunna hjälpa", sa han.
Nu när de har visat det automatiserade arbetsflödets potential hoppas Persson och teamet kunna införliva det i ett automatiserat laboratorium som analyserar NMR-data från tusentals eller till och med miljontals nya kemiska reaktioner åt gången.
Mer information: Maxwell C. Venetos et al, Deconvolution and Analysis of the 1H NMR Spectra of Crude Reaction Mixtures, Journal of Chemical Information and Modeling (2024). DOI:10.1021/acs.jcim.3c01864
Journalinformation: Journal of Chemical Information and Modeling
Tillhandahålls av Lawrence Berkeley National Laboratory