Forskare har utvecklat en plattform som kombinerar automatiserade experiment med AI för att förutsäga hur kemikalier kommer att reagera med varandra, vilket kan påskynda designprocessen för nya läkemedel.
Att förutsäga hur molekyler kommer att reagera är avgörande för upptäckten och tillverkningen av nya läkemedel, men historiskt sett har detta varit en trial-and-error-process, och reaktionerna misslyckas ofta. För att förutsäga hur molekyler kommer att reagera, simulerar kemister vanligtvis elektroner och atomer i förenklade modeller, en process som är beräkningsmässigt dyr och ofta inexakt.
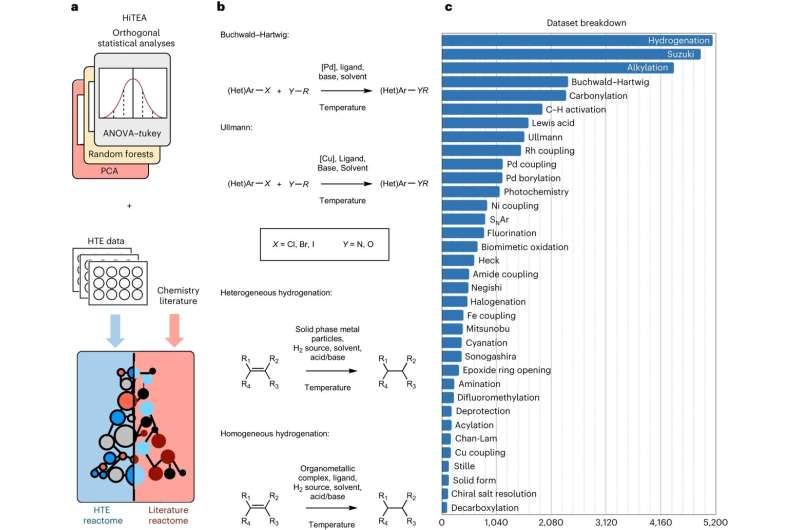
Nu har forskare från University of Cambridge utvecklat ett datadrivet tillvägagångssätt, inspirerat av genomik, där automatiserade experiment kombineras med maskininlärning för att förstå kemisk reaktivitet, vilket kraftigt påskyndar processen. De har kallat sitt tillvägagångssätt, som validerades på en datauppsättning med mer än 39 000 farmaceutiskt relevanta reaktioner, den kemiska "reaktomen."
Deras resultat, rapporterade i tidskriften Nature Chemistry , är resultatet av ett samarbete mellan Cambridge och Pfizer.
"Reaktomen kan förändra vårt sätt att tänka på organisk kemi", säger Dr Emma King-Smith från Cambridges Cavendish Laboratory, tidningens första författare. "En djupare förståelse för kemin skulle kunna göra det möjligt för oss att göra läkemedel och så många andra användbara produkter mycket snabbare. Men mer fundamentalt, den förståelse vi hoppas kunna generera kommer att vara till nytta för alla som arbetar med molekyler."
Reaktommetoden plockar ut relevanta korrelationer mellan reaktanter, reagenser och reaktionens prestanda från data, och pekar ut luckor i själva data. Data genereras från mycket snabba, eller hög genomströmning, automatiserade experiment.
"Kemi med hög genomströmning har förändrat spelet, men vi trodde att det fanns ett sätt att avslöja en djupare förståelse av kemiska reaktioner än vad som kan observeras från de initiala resultaten av ett experiment med hög genomströmning", säger King-Smith.
"Vårt tillvägagångssätt avslöjar de dolda sambanden mellan reaktionskomponenter och resultat", säger Dr Alpha Lee, som ledde forskningen. "Datauppsättningen som vi tränade modellen på är enorm – den kommer att hjälpa till att föra processen för kemisk upptäckt från trial-and-error till big data-åldern."
I en relaterad artikel, publicerad i Nature Communications , utvecklade teamet ett tillvägagångssätt för maskininlärning som gör det möjligt för kemister att introducera exakta transformationer till förspecificerade molekylregioner, vilket möjliggör snabbare läkemedelsdesign.
Tillvägagångssättet gör det möjligt för kemister att justera komplexa molekyler – som en designförändring i sista minuten – utan att behöva göra dem från grunden. Att göra en molekyl i labbet är vanligtvis en process i flera steg, som att bygga ett hus. Om kemister vill variera kärnan i en molekyl är det konventionella sättet att bygga om molekylen, som att slå ner huset och bygga om från grunden. Kärnvariationer är dock viktiga för medicindesign.
En klass av reaktioner som kallas funktionaliseringsreaktioner i sen skede försöker att direkt introducera kemiska omvandlingar till kärnan, vilket undviker behovet av att börja om från början. Det är dock utmanande att göra funktionalisering i sen skede selektiv och kontrollerad – det finns vanligtvis många regioner av molekylerna som kan reagera, och det är svårt att förutsäga resultatet.
"Sent skede funktionalisering kan ge oförutsägbara resultat och nuvarande metoder för modellering, inklusive vår egen expertintuition, är inte perfekta," sa King-Smith. "En mer prediktiv modell skulle ge oss möjlighet till bättre screening."
Forskarna utvecklade en maskininlärningsmodell som förutsäger var en molekyl skulle reagera, och hur reaktionsplatsen varierar som en funktion av olika reaktionsförhållanden. Detta gör det möjligt för kemister att hitta sätt att justera kärnan i en molekyl.
"Vi förtränade modellen på en stor mängd spektroskopiska data - som effektivt lärde modellen generell kemi - innan vi finjusterade den för att förutsäga dessa komplicerade transformationer", säger King-Smith. Detta tillvägagångssätt gjorde det möjligt för teamet att övervinna begränsningen av låga data:det finns relativt få funktionaliseringsreaktioner i sena stadier rapporterade i den vetenskapliga litteraturen. Teamet validerade modellen experimentellt på en mångsidig uppsättning läkemedelsliknande molekyler och kunde exakt förutsäga reaktivitetsställena under olika förhållanden.
"Tillämpningen av maskininlärning på kemi stryps ofta av problemet att mängden data är liten jämfört med det enorma kemiska utrymmet", säger Lee. "Vårt tillvägagångssätt – att designa modeller som lär sig av stora datamängder som liknar men inte samma som problemet vi försöker lösa – löser denna grundläggande utmaning med låg data och kan låsa upp framsteg bortom funktionalisering i sena skeden."
Mer information: Emma King-Smith et al, Undersöka den kemiska "reaktomen" med experimentdata med hög genomströmning, Nature Chemistry (2024). DOI:10.1038/s41557-023-01393-w
Predictive Minisci funktionalisering i sen skede med överföringsinlärning, naturkommunikation (2024). DOI:10.1038/s41467-023-42145-1. www.nature.com/articles/s41467-023-42145-1
Journalinformation: Nature Communications , Naturkemi
Tillhandahålls av University of Cambridge