Jianwei Shuai's team och Jiahuai Hans team vid Xiamen University har utvecklat en djup autoencoder-baserad dataoberoende datainsamlingsdataanalysmjukvara för proteinmasspektrometri, som realiserar analysen av relevanta peptider och proteiner från komplexa proteinmasspektrometridata, och visar överlägsenhet och metodens mångsidighet på olika instrument och artprover. Studien publicerades i Research som "Dear-DIA XMBD :djup autokodare för dataoberoende insamlingsproteomik".
Proteiner spelar en avgörande roll som utförare av cellulära livsaktiviteter och driver en myriad av avgörande biologiska processer. Följaktligen har området proteomik fått stor uppmärksamhet. Proteomics involverar en omfattande studie av proteinegenskaper, inklusive post-translationella modifieringar, proteinuttrycksnivåer, protein-protein-interaktioner och mer. Dess övergripande mål är att få en holistisk förståelse av sjukdomspatogenes, cellulär metabolism och andra vitala processer på proteinnivå.
Bland de viktigaste analytiska teknikerna inom proteomikforskning framstår proteinmasspektrometri som den mest kritiska. Med tiden har masspektrometritekniken utvecklats för att ge forskare tillförlitliga och dynamiska verktyg för proteomikanalys.
Två huvudsakliga tillvägagångssätt för proteinmasspektrometri är databeroende förvärv (DDA) och dataoberoende förvärv (DIA). I DDA förvärvas alla peptidprekursorjonspektra (MS1) i fullskanningsläge, följt av val av de mest N-intensiva peptidjonerna för fragmentering för att erhålla fragmentjonspektra (MS2).
Trots dess användbarhet står DDA inför utmaningar relaterade till experimentell reproducerbarhet och detektion av peptider med låg förekomst på grund av peptidfragmenteringens slumpmässighet och det föredragna urvalet av högintensiva peptider.
För att övervinna dessa begränsningar har DIA-insamlingsmetoden införts. Denna teknik delar upp mass-till-laddning-förhållandeintervallet för moderjonspektra i flera fönster och fragmenterar sekventiellt alla peptider inom varje fönster för att erhålla dotterjonspektra. En vanlig DIA-metod är Sequential Window Acquisition av alla teoretiska fragmentjoner (SWATH).
Medan DIA-insamlingsdata behåller mer omfattande proteomisk information, utgör dess stora datastorlek, höga dimensionalitet och komplexa spektrala signaler utmaningar för analysen. Som ett resultat av detta har DIA-datautvinning blivit ett stort fokus inom proteomiksamhället.
Jianwei Shuais team och Jiahuai Hans team samarbetade för att utveckla Dear-DIA, en djupinlärningsbaserad dataoberoende dataanalysmjukvara för förvärv, som realiserar identifieringen av fragmentjoner som motsvarar olika peptider från komplexa DIA-förvärvsspektra och visar generaliseringen till komplexa prover. från olika arter.
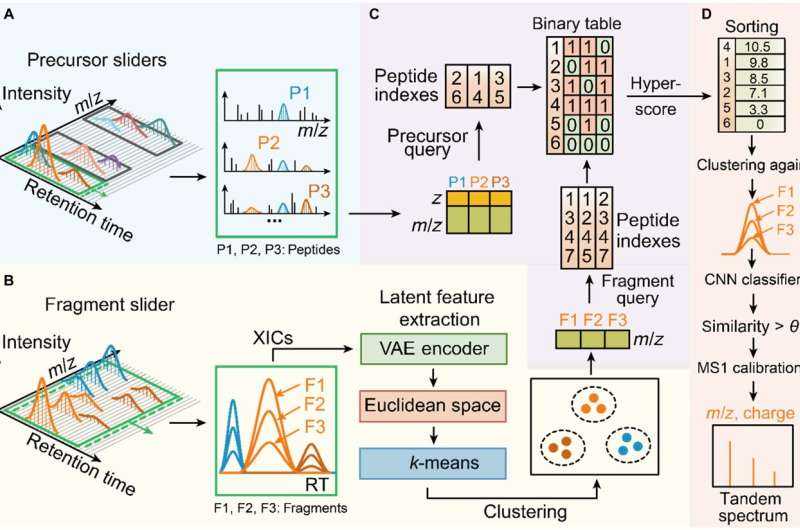
Dear-DIA delar först upp spektra i ett reglage med fast bredd med en fast bredd längs retentionstidsriktningen (RT), och varje reglage innehåller en uppsättning prekursorspektra MS1 och fragmentspektra MS2 som minsta bearbetningsenhet. Sedan användes en peak-finding-algoritm för att ta bort bakgrundsjonerna med låg signal-till-brus och behålla kandidat-prekursorjonerna och kandidatfragmentjonerna.
Därefter använder Dear-DIA en variationsautokodare för att extrahera toppegenskaperna för fragmentjoner och kartlägger särdragen i det euklidiska rymden, och grupperar sedan funktionerna med olika klasser av fragment som motsvarar olika peptider, vilket förverkligar spektrogramdekonvolutionsprocessen.
Dear-DIA inkluderar en indexeringsalgoritm som kallas PIndex, som matchar prekursorerna till fragmentens klustringsresultat och väljer de bästa parningsresultaten genom att poängsätta. Dear-DIA använder ett konvolutionellt neuralt nätverk för att räkna om toppformlikheten för fragment i samma klass för att eliminera störande joner och klustringsresultat med låg likhet.
Författarna testade först prestandan hos Dear-DIA på en SGS Human-datauppsättning innehållande 422 syntetiska peptider av stabila isotopmärkta standarder uppdelade i 10 utspädningsgradienter (från 1-faldig till 512-faldig utspädning), och DIA-data erhölls på en AB SCIEX TTOF5600 masspektrometer som använder SWATH-tekniken för att få DIA-data.
Analysresultaten visade att Dear-DIA hittade fler syntetiska peptider i alla utspädda lösningar jämfört med de två vanliga analytiska metoderna, Spectronaut 14 och DIA-Umpire. Författarna jämförde också antalet peptider och proteiner som hittats med de olika analysmetoderna för SGS Human och L929 Mouse datamängder. Resultaten visade att Dear-DIA kunde hitta fler peptider och proteiner jämfört med Spectronaut 14 och DIA-Umpire, vilket täckte mer än 85 % av deras resultat.
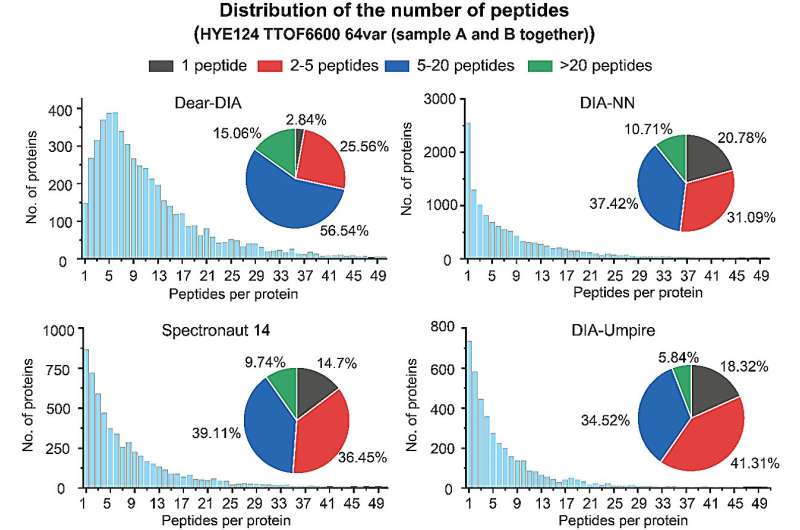
Tillförlitligheten hos proteomikanalysresultat kan också demonstreras av antalet peptider som identifierats för varje protein. Proteiner med 2 eller fler identifierade peptider anses generellt vara mer trovärdiga identifikationer. Författarna jämförde antalet proteiner kontra peptider som rapporterats av Dear-DIA med befintlig programvara på en datauppsättning av blandade arter (HYE124 TTOF6600 64var dataset).
Datauppsättningen innehåller proteiner från tre arter, människa, jäst och E. coli, och data förvärvades på en AB SCIEX TTOF6600 masspektrometer med användning av SWATH-metoden, med moderjonspektra innehållande 64 variabla fönster. Analysresultaten visade att 97,16 % av proteinerna som hittats av Dear-DIA kunde motsvara 2 och fler peptider, vilket är mycket högre än DIA-NN, Spectronaut 14 och DIA-Umpire.
Dataoberoende insamlingstekniker för proteomik har antagits i stor utsträckning, och relaterade analysalgoritmer har blivit en forskningshotspot. Proteinupptäckt från massiva masspektrometridata är en intressant och utmanande uppgift. I den här artikeln utvecklade teamet Dear-DIA, en analysmjukvara baserad på djupinlärning, som används för att bearbeta en mängd mycket komplexa DIA-insamlingsdata, och som kan upptäcka fler peptider och proteiner, förutom att reproducera de flesta av resultaten av Spectronaut och DIA-Umpire.
Dessutom, även om träningsdataset är från E. coli, visar den utmärkta prestandan hos Dear-DIA på datasetet för blandade arter dess starka generaliseringsförmåga att analysera komplexa proteomikdata. Deep learning, som ett allmänt använt verktyg för big data-analys, har visat utmärkta datautvinningsmöjligheter för att upptäcka djupa inneboende associationer i big data.
Användningen av djupinlärning för att analysera proteomikmasspektrometridata har stor potential och kommer ytterligare att främja studiet av grundläggande frågor som proteinsignaleringsnätverk.
Mer information: Qingzu He et al, Dear-DIA XMBD :Deep Autoencoder möjliggör dekonvolution av dataoberoende förvärvsproteomik, forskning (2023). DOI:10.34133/research.0179
Journalinformation: Forskning
Tillhandahålls av Research