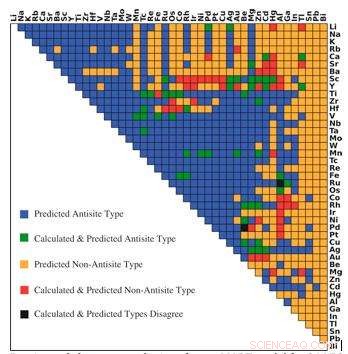
Förutsägelser av dominerande defekttyp från r-MART-modell för 946 B2-typ intermetallics. Färger indikerar förhållandet mellan förutsägelse och beräkningar som visas i förklaringen. Kredit:Bharat Medasani, Berkeley Lab / PNNL
För första gången, forskare vid Lawrence Berkeley National Laboratory (Berkeley Lab) har byggt och tränat maskininlärningsalgoritmer för att förutsäga defektbeteende i vissa intermetalliska föreningar med hög noggrannhet. Denna metod kommer att påskynda forskningen av nya avancerade legeringar och nya lättviktsmaterial för applikationer som sträcker sig från fordon till flyg och mycket mer.
Deras resultat publicerades i decembernumret 2016 av Naturens beräkningsmaterial .
Material är aldrig kemiskt rena och strukturellt felfria. De innehåller nästan alltid defekter, som spelar en viktig roll för att diktera egenskaper. Dessa defekter kan uppstå som lediga platser, som i huvudsak är "hål" i ämnets kristallstruktur, eller antisitedefekter, som i huvudsak är atomer placerade på fel kristallställe. Förståelse för sådana punktdefekter är avgörande för forskare som designar material eftersom de kan ha en dramatisk effekt på långvarig strukturell stabilitet och styrka.
Traditionellt, Forskare har använt en kvantmekanisk beräkningsmetod som kallas densitetsfunktionella beräkningar för att förutsäga vilka typer av defekter som kan bildas i en given struktur och hur de påverkar materialets egenskaper. Även om det är effektivt, detta tillvägagångssätt är mycket beräkningsmässigt dyrt att utföra för punktdefekter som begränsar omfattningen av sådana undersökningar.
"Densitetsfunktionella beräkningar fungerar bra om du modellerar en liten enhet, men om du vill göra din modelleringscell större ökar beräkningskraften som krävs för att göra detta avsevärt, " säger Bharat Medasani, en tidigare postdoc i Berkeley Lab och huvudförfattare till npj-tidningen. "Och eftersom det är beräkningsmässigt dyrt att modellera defekter i ett enda material, att göra den här typen av brute force-modellering för tiotusentals material är inte genomförbart."
För att övervinna dessa datorutmaningar, Medasani och hans kollegor utvecklade och tränade maskininlärningsalgoritmer för att förutsäga punktdefekter i intermetalliska föreningar, med fokus på den allmänt observerade B2-kristallstrukturen. Initialt, de valde ett urval av 100 av dessa föreningar från Materials Project Database och körde funktionella densitetsberäkningar på superdatorer vid National Energy Research Scientific Computing Center (NERSC), en DOE Office of Science User Facility vid Berkeley Lab, för att identifiera deras defekter.
Eftersom de hade ett litet dataprov att arbeta utifrån, Medasani och hans team använde en skogsmetod som kallas gradientförstärkning för att utveckla sin maskininlärningsmetod med hög noggrannhet. I detta tillvägagångssätt byggdes ytterligare maskininlärningsmodeller successivt och kombinerades med tidigare modeller för att minimera skillnaden mellan modellernas förutsägelser och resultaten från densitetsfunktionsberäkningar. Forskarna upprepade processen tills de uppnådde en hög nivå av noggrannhet i sina förutsägelser.
"Detta arbete är i grunden ett proof of concept. Det visar att vi kan köra funktionella densitetsberäkningar för några hundra material, träna sedan maskininlärningsalgoritmer för att exakt förutsäga punktdefekter för en mycket större grupp av material, " säger Medasani, som nu är postdoktor vid Pacific Northwest National Laboratory.
"Fördelen med detta arbete är att vi nu har en beräkningsmässigt billig maskininlärningsmetod som snabbt och exakt kan förutsäga punktdefekter i nya intermetalliska material", säger Andrew Canning, en Berkeley Lab Computational Scientist och medförfattare på npj-tidningen. "Vi behöver inte längre köra mycket kostsamma första principberäkningar för att identifiera defektegenskaper för varje ny metallförening."
"Det här verktyget gör det möjligt för oss att förutsäga metalldefekter snabbare och robust, vilket i sin tur kommer att påskynda materialdesign, säger Kristin Persson, en Berkeley Lab-forskare och chef för materialprojektet, ett initiativ som syftar till att drastiskt minska den tid som behövs för att uppfinna nytt material genom att tillhandahålla öppen webbaserad tillgång till beräknad information om känt och förutspått material. Som en förlängning av detta arbete har en Python-verktygssats med öppen källkod för modellering av punktdefekter i halvledare och isolatorer (PyCDT) utvecklats.