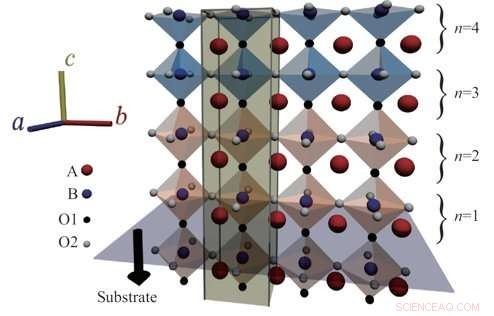
Fig. 1:Schematisk över perovskitoxidgränssnitt. Kredit:Osaka University
Perovskiter är en typ av mineral och klass av material, och har väckt stor uppmärksamhet för sina potentiella tillämpningar för teknologier som de som används i solceller. Dessa unika material har välordnade strukturer och visar många intressanta egenskaper som kan vara användbara inom andra områden av elektronik. En sådan mängd egenskaper i samma strukturella ryggrad tillåter olika typer av perovskiter, med olika egenskaper, att vara jämnt sammanfogade utan att bryta gallerkoherensen. Att kunna undersöka strukturerna vid dessa gränssnitt är viktigt för forskare som studerar perovskiter, men för närvarande använda tekniker har otillräcklig upplösning eller ger komplexa resultat som är mycket svåra att analysera.
Nu, Osaka University-ledda forskare har hittat ett sätt att modellera perovskitoxidgränssnitt med stor precision och noggrannhet med hjälp av en ny datoriserad metod för att plocka ut rätt struktur från röntgendata. De rapporterade nyligen sina fynd i Journal of Applied Crystallography.
"Att använda typisk scanningstransmissionselektronmikroskopi på perovskitoxider kräver att prover skärs, som kan skada ytan och påverka upplösningen, " Studiens huvudförfattare Masato Anada säger. "Röntgendiffraktionsmetoder för ytröntgen undviker dessa effekter, men att analysera data är komplicerat, så få människor använder denna metod. Vår Monte Carlo-baserade förfiningsmetod ger ett snabbt sätt att söka efter den mest sannolika strukturen från röntgendata, och är tillräckligt mångsidig för att kunna tillämpas på mer variabla gränssnitt."
Monte Carlo-metoder hjälper till att förutsäga hur strukturen av ett gränssnitt förmodligen ser ut. Genom att göra små förändringar, med vissa restriktioner, många olika möjliga strukturer kan simuleras slumpmässigt.
Genom att tillämpa denna teknik på gränssnittet mellan perovskiter och jämföra simulerade röntgendata med verkliga mätningar kan forskarna snabbt identifiera de mest troliga perovskitstrukturerna.
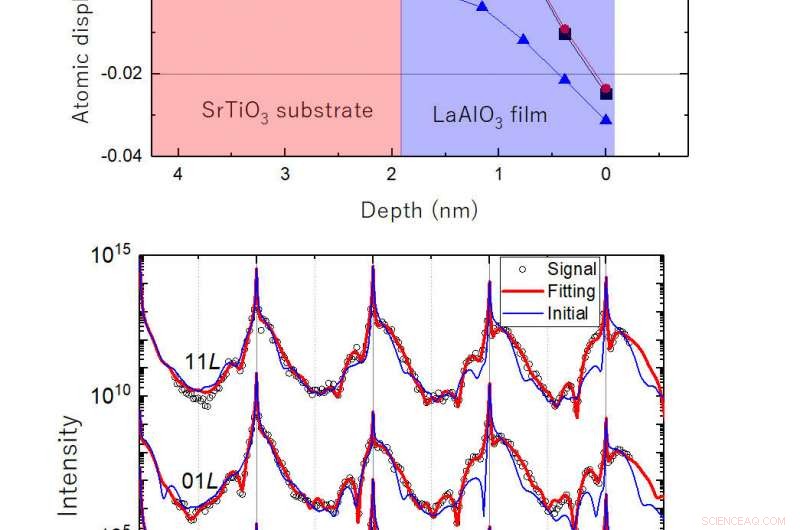
Fig.2:Exempel på mjukvarans prestanda. (överst) Atomförskjutning av modellstruktur som funktion av djup. (nederst) Spridda röntgenintensitetsprofiler beräknade från modellstrukturen (demo-data, öppna cirklar), initial strukturell modell (blå kurva) och resultatet av förfiningen (röd kurva). I denna figur, analysen av demo-data för att visa metodens noggrannhet. Analysen på ett experimentellt erhållet dataset redovisas också. Kredit:Osaka University
De testade sin nya metod på en simulerad röntgendatauppsättning från en realistisk gränssnittsstruktur mellan två typer av perovskitoxider, och den slutliga strukturen som förfinats av deras modellering var mycket nära den faktiska strukturen för gränssnittet.
"Funktioner i perovskite-gränssnitt är idealiska för att testa vissa teorier inom den kondenserade materiens fysik och för att göra nya typer av elektroniska materialsystem, " medförfattare Yusuke Wakabayashi säger. "Vårt tillvägagångssätt gör det mycket lättare att analysera komplexa strukturella data för dessa gränssnitt, och den är också robust för ojämna gränssnittsstrukturer. Detta tillvägagångssätt borde vara användbart för alla som för närvarande undersöker dessa strukturer."