Nya modeller för djupinlärning förutsäger interaktioner mellan atomer i organiska molekyler. Dessa modeller kommer att hjälpa beräkningsbiologer och läkemedelsutvecklingsforskare att förstå och behandla sjukdomar. Upphovsman:Los Alamos National Laboratory
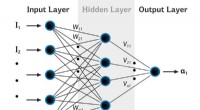
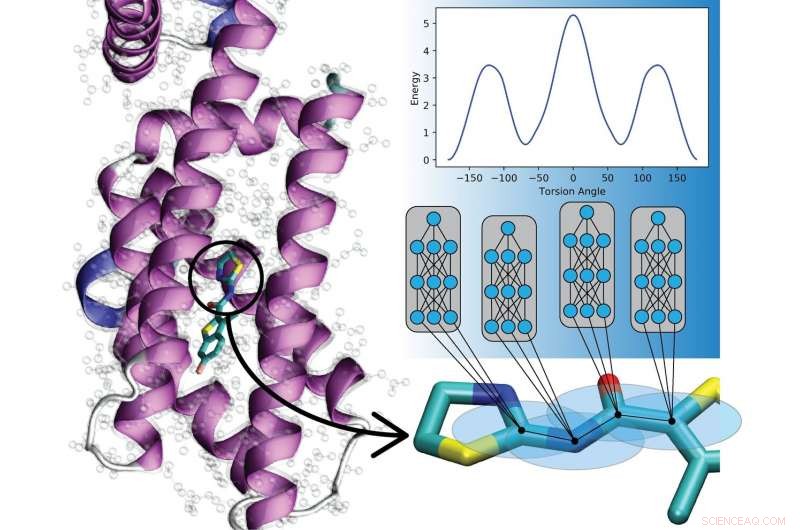
Nytt arbete från Los Alamos National Laboratory, University of North Carolina at Chapel Hill, och University of Florida visar att artificiella neurala nät kan tränas för att koda kvantmekaniska lagar för att beskriva molekylernas rörelser, överladdningssimuleringar potentiellt inom ett brett spektrum av områden.
"Detta betyder att vi nu kan modellera material och molekylär dynamik miljarder gånger snabbare jämfört med konventionella kvantmetoder, samtidigt som man behåller samma noggrannhetsnivå, sa Justin Smith, Los Alamos fysiker och Metropolis Fellow i laboratoriets teoretiska avdelning. Att förstå hur molekyler rör sig är avgörande för att utnyttja deras potentiella värde för läkemedelsutveckling, proteinsimuleringar och reaktiv kemi, till exempel, och både kvantmekanik och experimentella (empiriska) metoder ingår i simuleringarna.
Den nya tekniken, kallad ANI-1ccx-potentialen, lovar att främja kapaciteten hos forskare inom många områden och förbättra noggrannheten hos maskininlärningsbaserade potentialer i framtida studier av metallegeringar och detonationsfysik.
Kvantmekaniska (QM) algoritmer, används på klassiska datorer, kan exakt beskriva de mekaniska rörelserna för en förening i dess driftsmiljö. Men QM skalar mycket dåligt med olika molekylstorlekar, begränsar omfattningen av möjliga simuleringar kraftigt. Även en liten ökning av molekylstorleken inom en simulering kan dramatiskt öka beräkningsbördan. Så utövare använder ofta empirisk information, som beskriver atomernas rörelse i termer av klassisk fysik och Newtons lagar, möjliggör simuleringar som skala till miljarder atomer eller miljontals kemiska föreningar.
Traditionellt, empiriska potentialer har varit tvungna att göra en avvägning mellan noggrannhet och överförbarhet. När potentialens många parametrar är finjusterade för en förening, noggrannheten minskar på andra föreningar.
Istället, Los Alamos-laget, med University of North Carolina vid Chapel Hill och University of Florida, har utvecklat ett tillvägagångssätt för maskininlärning som kallas transfer learning som låter dem bygga empiriska potentialer genom att lära sig från data som samlats in om miljontals andra föreningar. Det nya tillvägagångssättet med den empiriska potentialen för maskininlärning kan appliceras på nya molekyler på millisekunder, möjliggör forskning om ett mycket större antal föreningar under mycket längre tidsperioder.