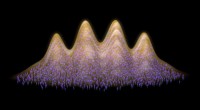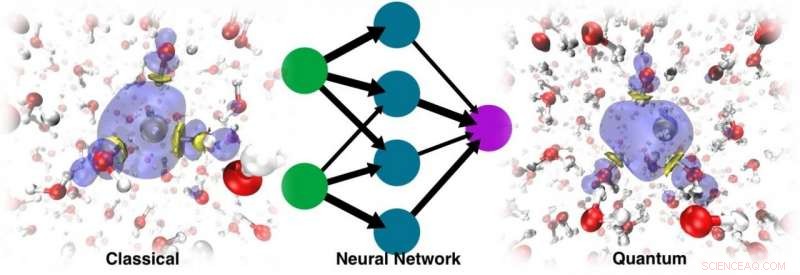
Dynamiken som kördes med den resulterande ML PES kunde inte bara återställa den stabila kaviteten, men kunde också spåra den korrekta lokaliseringsdynamiken Kredit:@Vladimir Rybkin
Beteendet hos den solverade elektronen e-aq har grundläggande konsekvenser för elektrokemi, fotokemi, högenergikemi, såväl som för biologi - dess icke-jämviktsprekursor är ansvarig för strålningsskador på DNA - och det har förståeligt nog varit ämnet för experimentell och teoretisk undersökning i mer än 50 år.
Även om den hydratiserade elektronen verkar vara enkel – den är den minsta möjliga anjonen såväl som det enklaste reduktionsmedlet inom kemin – är det svårt att fånga dess fysik. De är kortlivade och genereras i små mängder och så omöjliga att koncentrera och isolera. Deras struktur är därför omöjlig att fånga med direkt experimentell observation såsom diffraktionsmetoder eller NMR. Teoretisk modellering har visat sig vara lika utmanande.
Density functional theory (DFT) är den elektroniska strukturmetod som oftast används för att studera den solverade elektronen och vattnet. Standarddensitetsfunktioner lider dock av delokaliseringsfel, gör det omöjligt att modellera radikaler exakt. Rent vatten komplicerar DFT approximationer avsevärt, även om att välja rätt funktionalitet kan leda till acceptabla resultat jämfört med högnivåer för elektronisk struktur och värden som kan observeras genom experiment. En korrekt beskrivning av flytande vatten kan också uppnås med många kropps kvantkemimetoder, men de är extremt dyra.
Även om ett nyligen genomfört genombrott baserat på molekylär dynamik i pikosekunder som saknar motstycke i komplexitet och som kräver beräkningsresurser på gränsen för vad som är möjligt gav ett avgörande argument till förmån för en kavitetsstruktur för e-aq, det resulterade inte i andra nya insikter eller i en fullständig statistisk beskrivning. Omfattande karakterisering av systemets egenskaper kräver mycket längre tidsskalor, men att simulera kvantkärnor på denna nivå av elektronisk strukturteori är för närvarande bortom beräkningsräckvidd.
Det moderna sättet att kringgå detta problem innebär användning av maskininlärning. Att träna ett ML-kraftfält eller en potentiell energiyta (PES) baserat på ab initio-data möjliggör mycket längre MD-simuleringar eftersom kostnaden för att utvärdera sådana energier och krafter är nästan försumbar jämfört med den som är förknippad med elektroniska strukturberäkningar. Problemet är att den solvatiserade elektronen är en icke-typisk art. Den har ingen atomistisk formel, vilket utgör ett problem eftersom maskininlärning PES arbetar med atomistiska representationer.
I artikeln "Simulating the Ghost:Quantum Dynamics of the Solvatated Electron, "Universitetet i Zürich forskare Vladimir Rybkin, doktoranden Jinggang Lan och föreläsaren Marcella Iannuzzi kombinerade sin expertis inom elektronisk struktur och solvatiserade elektroner med kunskapen från EPFL-professorn Michele Ceriotti och hans tidigare Ph.D. studenter Venkat Kapil, nu forskare vid Cambridge University, och Piero Gasparotto, nu forskare på Empa, inom maskininlärning och kvantdynamik. Den där, med bidrag från andra kollegor, resulterade i tillämpningen av ML-metoden på data som erhållits från en kvantkemimetod för många kroppar känd som andra ordningens Møller-Plessets störningsteori (MP2), en metod som ger en exakt beskrivning av vatten, i alla fall, utan någon speciell behandling av överskottselektronen.
De blev förvånade när de upptäckte att modellen kunde lära sig närvaron av den solvatiserade elektronen som en faktor som förvrängde strukturen hos det rena flytande vattnet. Dynamiken som kördes med den resulterande ML PES kunde inte bara återställa den stabila kaviteten, men kunde också spåra den korrekta lokaliseringsdynamiken, utgående från den delokaliserade överskottselektronen som läggs till vattnet. I slutet, ML simulerade elektronen som en sorts "spökpartikel" som inte var explicit närvarande i modellen.
Detta gjorde det möjligt för forskarna att uppnå en tidsskala på flera hundra pikosekunder och samla in tillförlitlig statistik genom att köra många beräkningsmässigt billiga klassiska banor och beräkna vibrationsspektra, strukturer och diffusion. ML-metoden gjorde det också möjligt för dem att simulera kvantkärnorna snarare än klassiska kärnor med vägintegral molekyldynamik (PIMD). Denna teknik är åtminstone en storleksordning beräkningsmässigt dyrare än klassisk MD och kan inte utföras utan ML PES på en hög nivå av elektronisk strukturteori.
Med hänsyn till de nukleära kvanteffekterna levereras exakta vibrationsspektra, låta forskarna kvantifiera effekterna av dessa effekter - som redan visat sig vara mycket viktiga i överflödig elektronens avslappningsdynamik - på den hydrerade elektronen. Det avslöjade också transient diffusion, en ovanlig, sällsynt händelse som inte finns i den klassiska regimen. Även om icke-transient diffusion av den solvatiserade elektronen uppnås genom lösningsmedelsutbyte följt av gradvis förskjutning av "elektronmolnet" eller spinndensitetsfördelning, övergående diffusion är snarare ett hopp av spinndensiteten från den stabila kaviteten till den intilliggande.
Medan spökpartikelmetoden tillämpades här på den solvatiserade elektronen, det kan också tillämpas på exciterade tillstånd och kvasipartiklar som polaroner, öppna upp för nya möjligheter att förena elektronisk strukturteori på hög nivå med maskininlärning för att uppnå mycket exakta dynamiksimuleringar till ett rimligt pris.