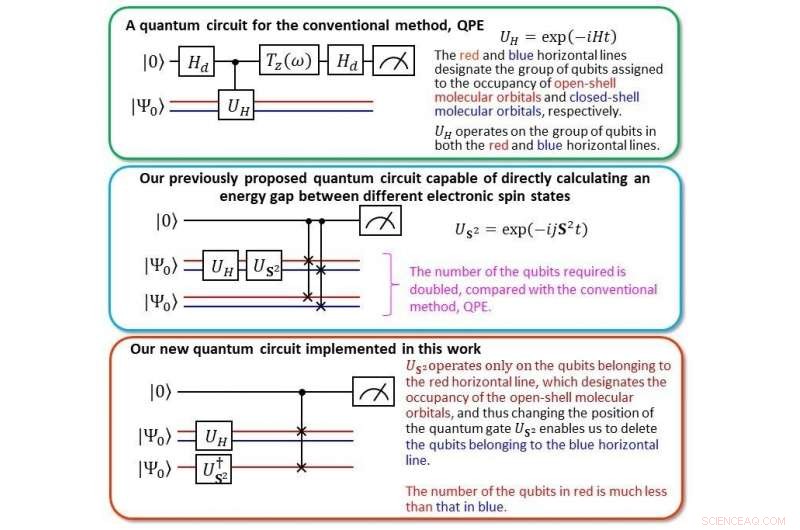
Jämförelse av den nya kvantkretsen med vår tidigare Credit:Kenji Sugisaki, Takeji Takui, Kazunobu Sato
Kvantdatorer har fått mycket uppmärksamhet den senaste tiden eftersom de förväntas lösa vissa problem som ligger utanför normala datorers kapacitet. Det primära till dessa problem är att bestämma de elektroniska tillstånden för atomer och molekyler så att de kan användas mer effektivt i en mängd olika industrier – från design av litiumjonbatterier till kiselteknik vid läkemedelsutveckling. Ett vanligt sätt som forskare har närmat sig detta problem är genom att beräkna de totala energierna för de individuella tillstånden för en molekyl eller atom och sedan bestämma skillnaden i energi mellan dessa tillstånd. I naturen, många molekyler växer i storlek och komplexitet, och kostnaden för att beräkna detta konstanta flöde är bortom kapaciteten för någon traditionell dator eller för närvarande etablera kvantalgoritmer. Därför, teoretiska förutsägelser av de totala energierna har bara varit möjliga om molekyler inte är stora och isolerade från sin naturliga miljö.
"För att kvantdatorer ska bli verklighet, dess algoritmer måste vara tillräckligt robusta för att exakt förutsäga de elektroniska tillstånden för atomer och molekyler, som de finns i naturen, " uppger Kenji Sugisaki och Takeji Takui från Graduate School of Science, Osaka City University.
I december 2020, Sugisaki och Takui, tillsammans med sina kollegor, ledde ett team av forskare att utveckla en kvantalgoritm som de kallar Bayesian eXchange kopplingsparameterkalkylator med Broken-symmetri wave functions (BxB), som förutsäger de elektroniska tillstånden för atomer och molekyler genom att direkt beräkna energiskillnaderna. De noterade att energiskillnader i atomer och molekyler förblir konstanta, oavsett hur komplexa och stora de blir trots att deras totala energier växer i takt med systemets storlek. "Med BxB, vi undvek den vanliga metoden att beräkna de totala energierna och riktade oss direkt mot energiskillnaderna, hålla beräkningskostnaderna inom polynomtiden, " säger de. "Sedan dess, vårt mål har varit att förbättra effektiviteten hos vår BxB-mjukvara så att den kan förutsäga atomernas och molekylernas elektroniska tillstånd med kemisk precision."
Genom att använda beräkningskostnaderna för en välkänd algoritm som kallas Quantum Phase Estimation (QPE) som riktmärke, "Vi beräknade de vertikala joniseringsenergierna för små molekyler som CO, O 2 , CN, F 2 , H 2 O, NH 3 inom 0,1 elektronvolts (eV) precision, säger laget, använder hälften av antalet qubits, vilket ger beräkningskostnaden i nivå med QPE.
Deras resultat kommer att publiceras online i marsupplagan av Journal of Physical Chemistry Letters .
Joniseringsenergi är en av de mest grundläggande fysikaliska egenskaperna hos atomer och molekyler och en viktig indikator för att förstå styrkan och egenskaperna hos kemiska bindningar och reaktioner. Kortfattat, att noggrant förutsäga joniseringsenergin gör att vi kan använda kemikalier utöver den nuvarande normen. Förr, det var nödvändigt att beräkna energierna för de neutrala och joniserade tillstånden, men med BxB-kvantalgoritmen, joniseringsenergin kan erhållas i en enda beräkning utan att inspektera de individuella totala energierna för de neutrala och joniserade tillstånden. "Från numeriska simuleringar av kvantlogikkretsen i BxB, vi fann att beräkningskostnaden för att läsa ut joniseringsenergin är konstant oavsett atomnummer eller molekylens storlek, " säger laget, "och att joniseringsenergin kan erhållas med en hög noggrannhet på 0,1 eV efter modifiering av längden på kvantlogikkretsen till att vara mindre än en tiondel av QPE."
Med utvecklingen av kvantdatorhårdvara, Sugisaki och Takui, tillsammans med sitt team, förväntar sig att BxB-kvantalgoritmen ska utföra högprecisionsenergiberäkningar för stora molekyler som inte kan behandlas i realtid med konventionella datorer.