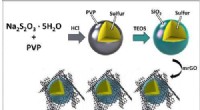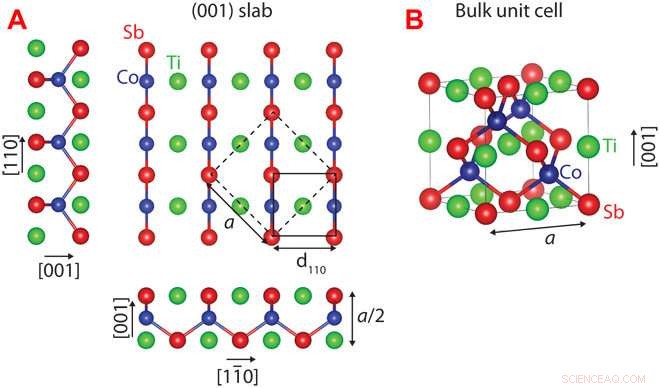
Kristallstruktur av CoTiSb. (A) Oavslappnad (001) platta av CoTiSb med TiSb -avslutning. Den konventionella bulkenhetscellen är markerad med streckade linjer (kantlängd a), och (1 × 1) ytenhetens cell markeras med en heldragen linje. (B) Konventionell kubisk enhetscell bestående av en CoSb zincblende -underdel som är fylld med Ti. Kreditera: Vetenskapliga framsteg (2018). DOI:10.1126/sciadv.aar5832
Vem skulle gissa att knäcka mysteriet om hur oändligt små atomer ordnar sig vid kanterna av kristaller i avancerade material kan vara så enkelt som en, två, tre?
För att modellera molekylstrukturen på en kristalls yta krävs normalt kraftfulla datorer, men University of Wisconsin – Madison ingenjörer har tagit fram en mycket enklare metod - en som är lika enkel som att räkna med penna och papper.
Den enkla strategin kan hjälpa till att åstadkomma ultrasnabba datachips baserade på annat material än kisel.
"Vi blev förvånade över att upptäcka att det var, faktiskt, så enkelt, "säger Jason Kawasaki, en professor i materialvetenskap och teknik från UW – Madison. "Med några ganska små tweaks, vi kunde förutsäga strukturer som var kvantitativt mycket exakta. "
De var så korrekta att hans nya förutsägelse, publicerad 1 juni, 2018 i tidningen Vetenskapliga framsteg , erbjuder ett snabbt och enkelt förfarande för att nollställa lovande material för användning i avancerad elektronik, till exempel kvantdatorer som löser problem mycket snabbare än konventionella kiselbaserade maskiner.
"Innan du kan använda material på intressanta sätt för nästa generations enheter, du måste förstå hur strukturen förändras på ytan, säger Kawasaki.
Noggrant att förutsäga kristallytstrukturer är ett problem som länge har bedrövade forskare. Atomer vid kanten av ett material tenderar att ordna om sig själva, ibland förlorar sina elektroniska eller magnetiska egenskaper.
Kawasaki och kollegor fokuserade på en typ av material som kallas halv-Heusler-föreningar, som har flera avstämbara elektroniska och magnetiska egenskaper. Tyvärr, många halv-Heuslers fungerar inte som förutsagt när de är ihopkopplade med andra material eller parade ner till en plan yta.
"När du har små omläggningar av atomer, du kan ha stora förändringar av fastigheter, säger Kawasaki.
Allt material består av atomer, som har kärnor i sina centrum omgiven av ständigt förskjutande moln av små subatomära partiklar som kallas elektroner. Atomer kan länka upp, eller bindning, genom att dela några av deras elektroner med varandra. Kristaller består av många atomer som är sammanbundna i ett regelbundet arrangerat och repetitivt mönster. Det mönstret går sönder, dock, vid kristallytor eller gränssnitt, lämnar några atomer utan partner och odelade elektroner dinglar bort från bulkmaterialet.
Inom kristallernas stela interiörer, sofistikerade simuleringar kan bestämma atomarrangemang, men datorer behöver initiala bästa gissningar på konfigurationer för att skapa strukturella förutsägelser.
Under en lång tid, bästa gissningar på ytan var omöjliga att komma på eftersom närvaron av dinglande elektroner får antalet möjliga konformationer att skjuta i höjden.
"Rätt verktyg och rätt teoretisk ram fanns inte, säger Kawasaki.
Den rätta teoretiska ramen visade sig vara förvånansvärt enkel, styrs av grundläggande kemiregler. Allt som behövs är att räkna upp alla elektroner som varje atom tar med sig till ytan, alla elektroner som förutspås vara i bindningar, och avgöra om dessa siffror matchar. När alla elektroner redovisas, strukturen är sannolikt stabil. Om inte, det är tillbaka till ritbordet.
Räkningen är så enkel att Kawasaki bokstavligen kan använda penna och papper för att utföra beräkningar.
Räkningsregler är kända för att fungera bra för enkla material. Dock, forskare antog att elektronmolnen för metallatomerna som utgör halva Heusler-material var för komplicerade för sådan grundläggande redovisning.
Kawasaki och kollegor visade att den uppfattningen var felaktig.
"Vi fann att många av de allmänna regler som har utvecklats för att förstå bindning i enkla system kan kartläggas till dessa mer komplexa material, säger Kawasaki.
Med denna metod, Kawasaki och kollegor förutspådde och bekräftade ytkonfigurationen för ett viktigt halv-Heusler-material som kallas kobolt titan antimon, som är en potentiellt användbar halvledare. Forskarna mätte kristallytan med avancerade bildtekniker, notera deras penna-och-papper förutsägelser stämde perfekt med riktiga atomkonfigurationer.
Forskarna tillämpade sedan sin metod på ytterligare två halva Heusler-föreningar, en halvmetall och en ferromagnet, och de planerar att identifiera mer lovande material.
Kawasaki utförde kristalltillväxt och mätningsexperiment i samarbete med Chris Palmstrøm, en fakultetsmedlem i el- och datorteknik och materialvetenskap vid University of California, Santa Barbara.