Cystisk fibros är en försvagande genetisk sjukdom som drabbar cirka 70 000 människor världen över. Nyligen har utvecklingen av en revolutionerande klass av läkemedel som kallas CFTR-modulatorer gett patienter nytt hopp genom att effektivt korrigera det felaktiga protein som är ansvarigt för sjukdomens symtom. Dessa läkemedel verkar på en molekylär nivå, och nu har banbrytande bilder fångat de intrikata detaljerna i deras interaktioner, vilket ger oöverträffade insikter om deras livsförändrande effekter.
I hjärtat av cystisk fibros ligger en mutation i CFTR-genen, som kodar för ett protein som är ansvarigt för att reglera rörelsen av salt och vatten över cellmembranen. Detta felaktiga protein gör att tjockt slem ansamlas i lungorna, vilket leder till kroniska luftvägsinfektioner, andningssvårigheter och en förkortad livslängd.
CFTR-modulatorer, såsom Ivacaftor, Lumacaftor och andra, fungerar genom att rikta och stabilisera CFTR-proteinet, rädda dess funktion och låta det fungera mer effektivt. Fram till nu har de exakta mekanismerna för hur dessa läkemedel interagerar med proteinet varit svårfångade.
Genom banbrytande avbildningstekniker som kryo-elektronmikroskopi och röntgenkristallografi har forskare framgångsrikt visualiserat de strukturella förändringar som induceras av CFTR-modulatorer på molekylär nivå. De resulterande bilderna avslöjar hur dessa läkemedel binder till specifika platser på CFTR-proteinet, vilket orsakar konformationsförändringar som i slutändan återställer dess korrekta funktion.
I fallet med Ivacaftor, till exempel, visar bilderna hur läkemedlet binder till en ficka i CFTR-proteinet och fungerar som ett "molekylärt lim" som stabiliserar dess struktur. Denna stabilisering leder till förbättrad kanal-gating, vilket möjliggör ett effektivt flöde av joner och vatten över cellmembranet.
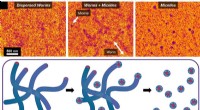
På liknande sätt avslöjar bilder av Lumacaftors interaktion med CFTR hur det hjälper till att korrigera proteinets veckning, vilket gör att det når sin funktionella form. Genom att åtgärda de underliggande strukturella defekterna förbättrar Lumacaftor proteinets förmåga att reglera passagen av joner och vatten.
Dessa banbrytande bilder fungerar som ett bevis på kraften i vetenskaplig innovation när det gäller att förstå sjukdomens molekylära grund och bana väg för riktade behandlingar. Beväpnade med denna kunskap kan forskare nu designa och utveckla mer effektiva CFTR-modulatorer och potentiellt utöka sina tillämpningar för att behandla andra genetiska sjukdomar som involverar felveckning av proteiner.
Resan för att reda ut de molekylära krångligheterna hos läkemedel mot cystisk fibros är långt ifrån över, men varje steg framåt ger nytt hopp för patienter som ivrigt väntar på livsförändrande behandlingar. När forskningen fortsätter att blomstra, har framtiden ett enormt löfte om att förbättra livet för otaliga individer som drabbats av denna förödande sjukdom.