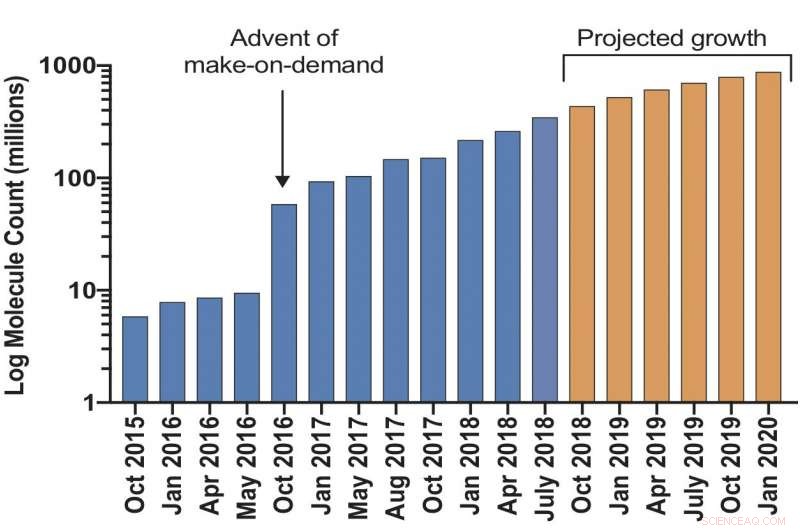
Ett virtuellt bibliotek av "make-on-demand"-molekyler som är tillgängliga för läkemedelsupptäckt förväntas överstiga 1 miljard föreningar nästa år. Kredit:Bryan Roth, M.D., Ph.D., vid University of North Carolina (UNC) Chapel Hill, Brian Shoichet, Ph.D., och John Irwin, Ph.D., vid University of California San Francisco, och kollegor.
Forskare har lanserat ett ultrastort virtuellt dockningsbibliotek som förväntas växa till mer än 1 miljard molekyler nästa år. Det kommer att utökas med 1000 gånger antalet sådana "tillverkade på begäran"-föreningar som är lätt tillgängliga för forskare för kemisk biologi och läkemedelsupptäckt. Ju större bibliotek, desto bättre är oddsen för att rensa bort inaktiva "lock"-molekyler som annars skulle kunna leda forskare nerför återvändsgränder. Projektet finansieras av National Institutes of Health.
"För att förbättra mediciner för psykiska sjukdomar, vi behöver screena ett stort antal potentiellt terapeutiska molekyler, " förklarade Joshua A Gordon, M.D., Ph.D., chef för NIH:s National Institute of Mental Health (NIMH), som medfinansierade forskningen. "Opartisk beräkningsmodellering tillåter oss att göra detta i en dator, avsevärt påskyndar processen att upptäcka nya behandlingar. Det gör det möjligt för forskare att praktiskt taget "se" en molekyl som dockar med dess receptorprotein - som ett fartyg i dess hamnplats eller en nyckel i dess lås - och förutsäga dess farmakologiska egenskaper, baserat på hur de molekylära strukturerna förutsägs interagera. Endast de relativt få kandidatmolekyler som bäst matchar målprofilen på datorn behöver göras fysiskt och testas i ett vått labb."
Bryan Roth, M.D., Ph.D., vid University of North Carolina (UNC) Chapel Hill, Brian Shoichet, Ph.D., och John Irwin, Ph.D., vid University of California San Francisco, och kollegor, rapportera om sina resultat 6 februari, 2019 i tidningen Natur . Studien fick stöd, till viss del, genom bidrag från NIMH, National Institute of General Medical Sciences (NIGMS), NIH Common Fund, och National Institute of Neurological Disorders and Stroke (NINDS).
NIH Common Funds Illuminating the Drugable Genome (IDG) Program – som lanserades 2014 för att katalysera forskning om proteiner som för närvarande är understuderade och potentiella mål för terapeutisk intervention – finansierade expansionen av dockningsbiblioteket.
Under de senaste åren, Roth, Shoichet, och kollegor har använt sin virtuella strukturbaserade dockningsmetod för att avslöja molekylära hemligheter för ett antipsykotiskt läkemedel och LSD dockade i sina respektive målreceptorer – och för att skapa ett designat smärtstillande medel som selektivt riktar sig mot hjärnans smärtstillande kretsar utan biverkningar av morfin.
Ett svindlande antal potentiella läkemedelsliknande molekyler är kända för att existera. Än, hundratals miljoner till miljarder olika molekyler har förblivit otillgängliga på grund av begränsningar av befintliga metoder som används för att sammanställa molekylära bibliotek, säger forskarna. Till exempel, deras virtuella strukturbaserade dockningsteknik, samtidigt som de lovar, riskerar att hitta många falska positiva eller "lockor – brister i modellen tillåter molekyler som verkar rimliga men som visar sig vara biologiskt inaktiva.
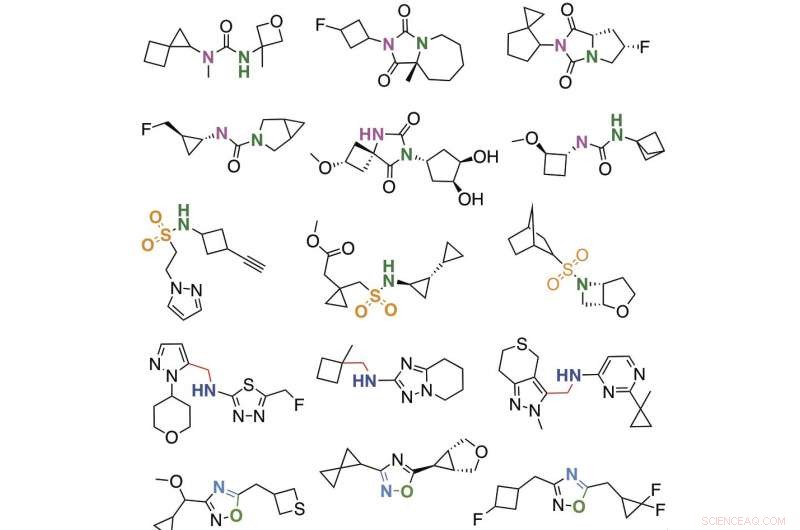
Urval av molekyler som upptäckts med hjälp av megadockningsbiblioteket. Kredit:Bryan Roth, M.D., Ph.D., vid University of North Carolina (UNC) Chapel Hill, Brian Shoichet, Ph.D., och John Irwin, Ph.D., vid University of California San Francisco, och kollegor.
För att övervinna denna utmaning, forskarna fokuserade på molekyler som är resultatet av 130 välkarakteriserade kemiska reaktioner med 70, 000 olika kemiska byggstenar. Datorsimuleringar med dessa molekyler visade att när storleken på ett bibliotek växte, förhållandet mellan "verkliga aktiva" och lockbeten ökade – precis som en studies statistiska kraft ökar med ett större urval.
I den nya studien, forskarna undersökte den strukturbaserade dockningen av 138 miljoner molekyler med antingen D4-receptorn, ett nyckelprotein som förmedlar handlingar av hjärnans kemiska budbärare dopamin, eller enzymet AmpC, som ger resistens mot vissa antibiotika och har visat sig vara svår att blockera.
"D4-receptorn är av särskilt intresse för NIMH på grund av dess roll i kognition och andra verkställande funktioner i hjärnans prefrontala cortex som ofta störs vid psykiska sjukdomar, sa Laurie Nadler, Ph.D., vid NIMH-avdelningen för neurovetenskap och grundläggande beteendevetenskap, programansvarig för anslaget som stöder D4-receptorstudien.
Forskarna syntetiserade och testade sedan, i ett labb, de 549 bästa molekylerna som praktiskt taget dockade bäst med D4-receptorn och 44 molekyler som dockade bäst med enzymet. Dessa studier avslöjade flera nya läkemedelsliknande molekyler som bara binder till D4-receptorn (och inte de närbesläktade D2- eller D3-dopaminreceptorerna) och slog på eller av receptorn. Dessutom, en molekyl (4163) framträdde som det mest potenta bindemedlet för AmpC någonsin. En virtuell molekyls dockningsgrad förutspådde dess faktiska sannolikhet att binda till D4-dopaminreceptorn i en labbanalys.
Upptäckten av nya och potenta molekyler för båda målen bekräftade också att ultrastora bibliotek innehåller molekyler som är bättre lämpade för en given receptorstruktur än mindre bibliotek och att virtuell dockning kan känna igen molekylerna och förutsäga det totala antalet förväntade aktiva föreningar inom ett bibliotek.
"Denna nya studie illustrerar potentialen hos opartisk beräkningsscreening och molekylär dockning för att upptäcka nya verktygsmolekyler och potentiella terapeutiska medel, tillhandahålla en snabb och robust väg som leder direkt till nya läkemedelsbehandlingar för psykiska sjukdomar, tillade Gordon.