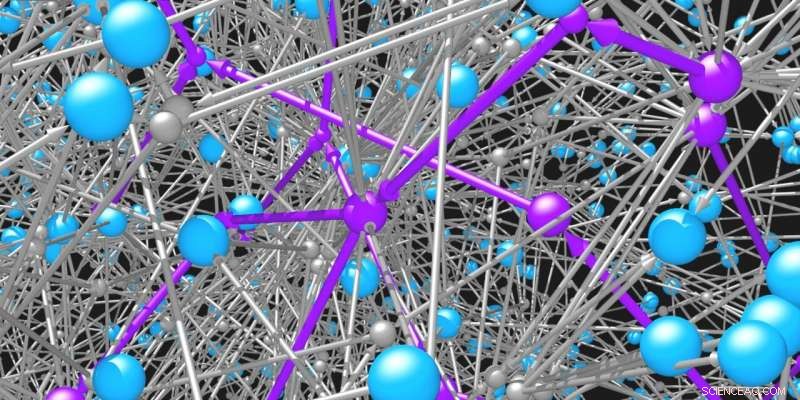
Molekyler (blå sfärer) är kopplade till varandra genom reaktionerna (grå sfärer och pilar) som de deltar i. Nätverket av möjliga organiska molekyler och reaktioner är omöjligt stort. Intelligenta sökalgoritmer behövs för att identifiera genomförbara vägar (lila) för att syntetisera önskade molekyler. Upphovsman:Mikolaj Kowalik &Kyle Bishop/Columbia Engineering
Forskare, från biokemister till materialvetare, har länge förlitat sig på den stora variationen av organiska molekyler för att lösa pressande utmaningar. Vissa molekyler kan vara användbara vid behandling av sjukdomar, andra för att tända våra digitala skärmar, ytterligare andra för pigment, målar, och plast. Varje molekyls unika egenskaper bestäms av dess struktur - det vill säga genom anslutningen av dess ingående atomer. När väl en lovande struktur har identifierats, det återstår den svåra uppgiften att göra den riktade molekylen genom en sekvens av kemiska reaktioner. Men vilka?
Organiska kemister arbetar i allmänhet bakåt från målmolekylen till utgångsmaterialen med hjälp av en process som kallas retrosyntetisk analys. Under denna process, kemisten står inför en rad komplexa och inbördes relaterade beslut. Till exempel, av tiotusentals olika kemiska reaktioner, vilken ska du välja för att skapa målmolekylen? När det beslutet är taget, du kan hitta dig själv med flera reaktantmolekyler som behövs för reaktionen. Om dessa molekyler inte är tillgängliga att köpa, hur väljer du då lämpliga reaktioner för att producera dem? Att intelligent välja vad som ska göras vid varje steg i denna process är avgörande för att navigera det stora antalet möjliga vägar.
Forskare vid Columbia Engineering har utvecklat en ny teknik baserad på förstärkningslärande som tränar en neural nätverksmodell för att korrekt välja den "bästa" reaktionen vid varje steg i den retrosyntetiska processen. Denna form av AI ger en ram för forskare att designa kemiska synteser som optimerar användarspecifika mål, t.ex. synteskostnader, säkerhet, och hållbarhet. Det nya tillvägagångssättet, publicerad 31 maj av ACS Central Science , är mer framgångsrik (med ~ 60%) än befintliga strategier för att lösa detta utmanande sökproblem.
"Förstärkningsinlärning har skapat datorspelare som är mycket bättre än människor på att spela komplexa videospel. Kanske är retrosyntes inte annorlunda! Denna studie ger oss hopp om att förstärkningsinlärningsalgoritmer kanske en dag är bättre än mänskliga spelare vid" spelet " retrosyntes, "säger Alán Aspuru-Guzik, professor i kemi och datavetenskap vid University of Toronto, som inte var inblandad i studien.
Teamet inramade utmaningen med retrosyntetisk planering som ett spel som schack och Go, där det kombinatoriska antalet möjliga val är astronomiskt och värdet av varje val osäkert tills syntesplanen är klar och dess kostnad utvärderas. Till skillnad från tidigare studier som använde heuristiska poängfunktioner - enkla tumregler - för att styra retrosyntetisk planering, denna nya studie använde förstärkningslärningstekniker för att göra bedömningar baserade på den neurala modellens egen erfarenhet.
"Vi är de första som tillämpar förstärkningslärande på problemet med retrosyntetisk analys, "säger Kyle Bishop docent i kemiteknik. "Utgående från ett tillstånd av fullständig okunnighet, där modellen absolut ingenting vet om strategi och tillämpar reaktioner slumpmässigt, modellen kan öva och öva tills den hittar en strategi som överträffar en mänskligt definierad heurist. "
I deras studie, Biskops team fokuserade på att använda antalet reaktionssteg som mätning av vad som gör en "bra" syntetisk väg. De fick sin förstärkande inlärningsmodell att skräddarsy sin strategi med detta mål i åtanke. Med hjälp av simulerad erfarenhet, laget utbildade modellens neurala nätverk för att uppskatta den förväntade synteskostnaden eller värdet för en given molekyl baserat på en representation av dess molekylära struktur.
Teamet planerar att utforska olika mål i framtiden, till exempel, träna modellen för att minimera kostnaderna snarare än antalet reaktioner, eller för att undvika molekyler som kan vara giftiga. Forskarna försöker också minska antalet simuleringar som krävs för att modellen ska lära sig dess strategi, eftersom utbildningsprocessen var ganska beräknande dyr.
"Vi förväntar oss att vårt retrosyntesspel snart kommer att följa schacks och Go's väg, där självlärda algoritmer konsekvent överträffar mänskliga experter, "Biskop noterar." Och vi välkomnar tävling. Som med schack-datorprogram, konkurrens är motorn för förbättringar av den senaste tekniken, och vi hoppas att andra kan bygga vidare på vårt arbete för att visa ännu bättre prestanda. "
Studien har titeln "Att lära sig retrosyntetisk planering genom simulerad erfarenhet."