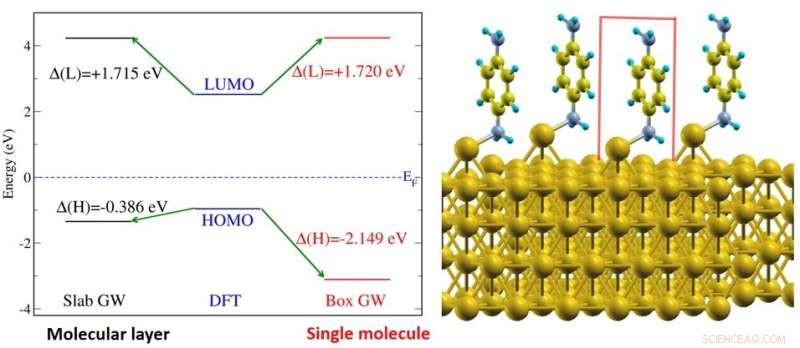
(Vänster) Figur visar elektronenerginivåjustering av bensen-diaminmolekyler på guldytesystem (visas till höger). Energinivåerna visas för ett molekylärt lager (svart) och för en enstaka molekyl (rött). (Höger) Illustration av bensen-diaminmolekylerna på guldytan. Kredit:National University of Singapore
NUS-fysiker har funnit att komplexa elektron-elektroninteraktioner förändrar energinivåerna vid molekyl-metall-gränssnitt, påverkar prestandan hos molekylära elektroniska enheter.
Molekylär elektronik innebär användning av molekyler som huvudbyggsten för att skapa de elektroniska kretsarna. Det kan potentiellt användas för att utveckla kretsar som är mycket mindre än de som är gjorda av konventionella kiselprocesser. Förstå de elektroniska egenskaperna hos gränssnittet mellan molekylerna och metallledarna, särskilt deras associerade energinivåer, är viktigt för att rationalisera och optimera enhetens prestanda. Detta är centralt för utvecklingen av molekylär elektronik.
En grundläggande egenskap hos varje molekyl är dess energigap, definieras som energiskillnaden mellan den högsta och lägsta orbitalenerginivån som är upptagen respektive oupptagen av elektroner. Dessa nivåer är också de viktigaste energinivåerna för enhetens prestanda. Energigapet hos en molekyl blir mindre när molekylen förs nära en metallyta; detta kommer att göra det lättare för laddningsbärare att röra sig mellan molekylen och metallkontakten. Denna förändring i gapet orsakas främst av elektroniska skärmningseffekter från metallytan, och kan vara så stor som flera elektronvolt. Dock, denna elektroniska screeningseffekt saknas i majoriteten av teoretiska studier om detta ämne.
En forskargrupp ledd av Prof Su Ying QUEK, från institutionen för fysik, NUS har belyst gränssnittets elektroniska strukturegenskaper för ett antal olika molekyler på guldytor med hjälp av state-of-the-art teoretiska och beräkningsmetoder som explicit tar hänsyn till elektroniska screeningseffekter från första principer. Forskarna utförde beräkningar på molekylära system förankrade av vanliga kemiska funktionella grupper (amin, pyridin- och tiolatgrupper). Forskargruppen fann att för en enda molekyl, den elektroniska screeningseffekten kan exakt förutsägas från en bildladdningsmodell, även i närvaro av kemiska bindningar. Bildladdningsmodellen är en klassisk elektrostatisk metod som approximerar den elektroniska screeningen av en testladdning med en bildladdning i metallen. Dock, i enheter med många molekyler, forskarna fann betydande ytterligare elektroniska screeningmekanismer. Förutom intermolekylära screeningseffekter, Substratmedierade intermolekylära interaktioner har också visat sig bidra till dessa ytterligare screeningsmekanismer. Resultaten tyder på att laddningsbärare lättare kan tunnla över gränssnittet i enheter med många molekyler.
Prof Quek sa, "Detta arbete ger värdefulla insikter om de många elektroneffekterna vid gränssnitten mellan molekyl och metall som involverar kemiska bindningar. Resultaten och resultaten från denna forskning utgör ett viktigt steg mot förståelsen och manipulationen av funktionella organiska system i utvecklingen av molekylära enheter."