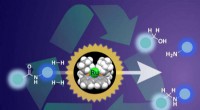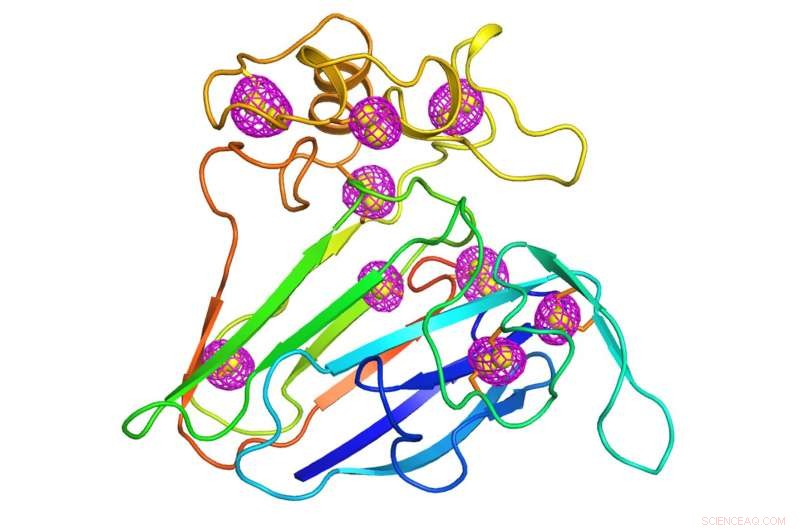
En tecknad serie som representerar strukturen av ett väl studerat växtprotein som fungerade som ett testfall för den nyutvecklade mikrokristallografitekniken. Magenta nätmönster som omger svavelatomer som är inneboende i proteinet (gula sfärer) indikerar de anomala signalerna som extraherades med hjälp av lågenergiröntgendiffraktion av tusentals kristaller som mäter mindre än 10 miljondelar av en meter, storleken på en bakterie. Kredit:Brookhaven National Laboratory
Att använda röntgenstrålar för att avslöja proteiners 3D-strukturer i atomskala har lett till otaliga framsteg när det gäller att förstå hur dessa molekyler fungerar i bakterier, virus, växter, och människor – och har väglett utvecklingen av precisionsläkemedel för att bekämpa sjukdomar som cancer och AIDS. Men många proteiner kan inte odlas till kristaller som är tillräckligt stora för att deras atomarrangemang ska kunna dechiffreras. För att tackla denna utmaning, forskare vid US Department of Energy's (DOE) Brookhaven National Laboratory och kollegor vid Columbia University har utvecklat en ny metod för att lösa proteinstrukturer från små kristaller.
Metoden bygger på unik provhantering, signalextraktion, och datasammanställningsmetoder, och en strållinje som kan fokusera intensiva röntgenstrålar vid Brookhavens National Synchrotron Light Source II (NSLS-II) – en DOE Office of Science-användaranläggning – till en miljondelsmetersplats, ungefär en femtiodel av ett människohårs bredd.
"Vår teknik öppnar verkligen dörren till att hantera mikrokristaller som tidigare varit otillgängliga, inklusive svårkristalliserade cellytereceptorer och andra membranproteiner, flexibla proteiner, och många komplexa mänskliga proteiner, " sa Brookhaven Lab-forskaren Qun Liu, motsvarande författare på studien, som publicerades den 3 maj, 2019, i IUCrJ , en tidskrift från International Union of Crystallography.
Dechiffrera proteinstrukturer
Proteinkristallografi har varit en dominerande metod för att lösa proteinstrukturer sedan 1958, förbättras med tiden eftersom röntgenkällor har blivit kraftfullare, möjliggör mer exakta strukturbestämningar. För att bestämma en proteinstruktur, forskare mäter hur röntgenstrålar som de som genereras vid NSLS-II diffrakterar, eller studsa av, atomerna i ett ordnat kristallint gitter som består av många kopior av samma proteinmolekyl, alla arrangerade på samma sätt. Diffraktionsmönstret förmedlar information om var atomerna finns. Men det är inte tillräckligt.
"Endast amplituderna för diffrakterade röntgenvågor registreras på detektorn, men inte deras faser (tidpunkten mellan vågorna), ", sa Liu. "Båda krävs för att rekonstruera en 3D-struktur. Detta är det så kallade kristallografiska fasproblemet."
Kristallografer har löst detta problem genom att samla in fasdata från en annan typ av spridning, känd som anomal spridning. Anomal spridning uppstår när atomer tyngre än ett proteins huvudkomponenter av kol, väte, och kväve absorberar och återutsänder en del av röntgenstrålarna. Detta händer när röntgenenergin är nära den energi som de tunga atomerna gillar att absorbera. Forskare infogar ibland på konstgjord väg tunga atomer som selen eller platina i proteinet för detta ändamål. Men svavelatomer, som förekommer naturligt i hela proteinmolekyler, kan också producera sådana signaler, om än svagare. Även om dessa onormala signaler är svaga, en stor kristall har vanligtvis tillräckligt många kopior av proteinet med tillräckligt med svavelatomer för att göra dem mätbara. Det ger forskarna den fasinformation som behövs för att lokalisera svavelatomerna och översätta diffraktionsmönstren till en fullständig 3D-struktur.
"När du väl känner till svavelpositionerna, du kan beräkna faserna för de andra proteinatomerna eftersom förhållandet mellan svavlet och de andra atomerna är fixerat, " sa Liu.
Men små kristaller, per definition, har inte så många kopior av proteinet av intresse. Så istället för att leta efter diffraktions- och fasinformation från upprepade kopior av ett protein i en enda stor kristall, Brookhaven/Columbia-teamet utvecklade ett sätt att ta mätningar från många små kristaller, och sedan samla ihop de samlade uppgifterna.
Små kristaller, stora resultat
För att hantera de små kristallerna, teamet utvecklade provgaller mönstrade med brunnar i mikrostorlek. Efter att ha hällt lösningsmedel som innehåller mikrokristallerna över dessa välmonterade galler, forskarna tog bort lösningsmedlet och frös kristallerna som fångades på gallren.
"Vi har fortfarande en utmaning, fastän, eftersom vi inte kan se var de små kristallerna finns på vårt rutnät, " sa Liu. "För att ta reda på det, vi använde mikrodiffraktion vid NSLS-II:s Frontier Microfocusing Macromolecular Crystallography (FMX) strållinje för att övervaka hela nätet. Skanna rad för rad, vi kan hitta var dessa kristaller är gömda."
Som Martin Fuchs, den ledande strållinjeforskaren på FMX, förklarade, "FMX-strållinjen kan fokusera hela intensiteten av röntgenstrålen ner till en storlek av en mikron, eller miljondels meter. Vi kan finkontrollera strålstorleken för att matcha den med storleken på kristallerna - fem mikron i fallet med det aktuella experimentet. Dessa förmågor är avgörande för att få den bästa signalen, " han sa.
Wuxian Shi, en annan FMX strållinjeforskare, noterade att "data som samlats in i rutnätsundersökningen innehåller information om kristallernas placering. Dessutom, vi kan också se hur väl varje kristall diffrakterar, vilket gör att vi bara kan välja de bästa kristallerna för datainsamling."
Forskarna kunde sedan manövrera provhållaren för att placera varje kartlagd mikrokristall av intresse tillbaka i mitten av precisionsröntgenstrålen för datainsamling.
De använde den lägsta tillgängliga energin vid strållinjen – inställd för att närma sig svavelatomernas absorptionsenergi så nära som möjligt – och samlade in onormal spridningsdata.
"De flesta kristallografiska strållinjer kunde inte nå svavelabsorptionskanten för optimerade anomala signaler, " sa medförfattaren Wayne Hendrickson vid Columbia University. "Lyckligtvis, NSLS-II är en världsledande synkrotronljuskälla som ger ljusa röntgenstrålar som täcker ett brett spektrum av röntgenenergi. Och även om vår energinivå låg något över den ideala absorptionsenergin för svavel, det genererade de onormala signaler vi behövde."
Men forskarna hade fortfarande en del arbete att göra för att extrahera dessa viktiga signaler och sammanställa data från många små kristaller.
"Vi får faktiskt tusentals bitar av data, " sa Liu. "Vi använde cirka 1400 mikrokristaller, var och en med sin egen datamängd. Vi måste sätta ihop all data från dessa mikrokristaller."
They also had to weed out data from crystals that were damaged by the intense x-rays or had slight variations in atomic arrangements.
"A single microcrystal does not diffract x-rays sufficiently for structure solution prior to being damaged by the x-rays, " said Sean McSweeney, deputy photon division director and program manager of the Structural Biology Program at NSLS-II. "This is particularly true with crystals of only a few microns, the size of about a bacterial cell. We needed a way to account for that damage and crystal structure variability so it wouldn't skew our results."
They accomplished these goals with a sophisticated multi-step workflow process that sifted through the data, discarded outliers that might have been caused by radiation damage or incompatible crystals, and ultimately extracted the anomalous scattering signals.
"This is a critical step, " said Liu. "We developed a computing procedure to assure that only compatible data were merged in a way to align the individual microcrystals from diffraction patterns. That gave us the required signal-to-noise ratios for structure determination."
Applying the technique
This technique can be used to determine the structure of any protein that has proven hard to crystallize to a large size. These include cell-surface receptors that allow cells of advanced lifeforms such as animals and plants to sense and respond to the environment around them by releasing hormones, transmitting nerve signals, or secreting compounds associated with cell growth and immunity.
"To adapt to the environment through evolution, these proteins are malleable and have lots of non-uniform modifications, " said Liu. "It's hard to get a lot of repeat copies in a crystal because they don't pack well."
I människor, receptors are common targets for drugs, so having knowledge of their varied structures could help guide the development of new, more targeted pharmaceuticals.
But the technique is not restricted to just small crystals.
"The method we developed can handle small protein crystals, but it can also be used for any size protein crystals, any time you need to combine data from more than one sample, " sa Liu.