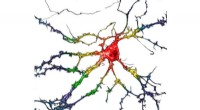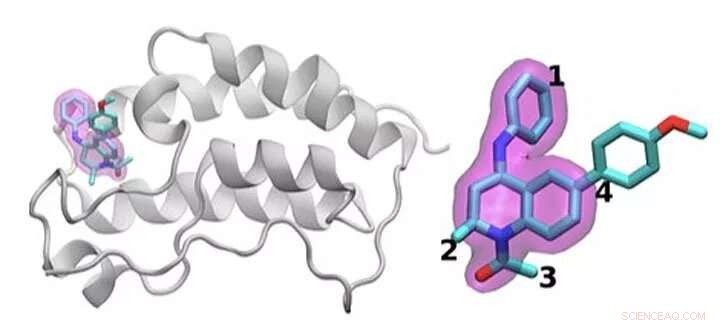
En schematisk bild av BRD4 -proteinet bundet till ett av 16 läkemedel baserat på samma tetrahydrokinolin -ställning (markerad i magenta). Regioner som är kemiskt modifierade mellan de läkemedel som undersöks i denna studie är märkta med 1 till 4. Typiskt endast en liten förändring görs i den kemiska strukturen från ett läkemedel till nästa. Detta konservativa tillvägagångssätt tillåter forskare att utforska varför ett läkemedel är effektivt medan ett annat inte är det. Upphovsman:Brookhaven National Laboratory
Att identifiera den optimala läkemedelsbehandlingen är som att träffa ett rörligt mål. För att stoppa sjukdomen, småmolekylära läkemedel binder tätt till ett viktigt protein, blockerar dess effekter i kroppen. Även godkända läkemedel fungerar vanligtvis inte hos alla patienter. Och med tiden, smittämnen eller cancerceller kan mutera, vilket gör ett en gång effektivt läkemedel värdelöst.
Ett kärnfysiskt problem ligger till grund för alla dessa frågor:optimering av interaktionen mellan läkemedelsmolekylen och dess proteinmål. Variationerna i läkemedelskandidatmolekyler, mutationsintervallet i proteiner och den övergripande komplexiteten hos dessa fysiska interaktioner gör detta arbete svårt.
Shantenu Jha vid Department of Energy's (DOE) Brookhaven National Laboratory och Rutgers University leder ett team som försöker effektivisera beräkningsmetoder så att superdatorer kan ta på sig en del av denna enorma arbetsbelastning. De har hittat en ny strategi för att ta itu med en del:att skilja hur läkemedelskandidater interagerar och binder med ett riktat protein.
För deras arbete, Jha och hans kollegor vann förra årets IEEE International Scalable Computing Challenge (SCALE) -pris, som känner igen skalbara datalösningar för verkliga vetenskapliga och tekniska problem.
För att designa ett nytt läkemedel, ett läkemedelsföretag kan börja med ett bibliotek med miljontals kandidatmolekyler som de begränsar till tusentals som visar någon initial bindning till ett målprotein. Att förfina dessa alternativ till ett användbart läkemedel som kan testas hos människor kan innebära omfattande experiment för att lägga till eller subtrahera atomgrupper på viktiga platser på molekylen och testa hur var och en av dessa förändringar förändrar hur den lilla molekylen och proteinet interagerar.
Simuleringar kan hjälpa till med denna process. Större, snabbare superdatorer och alltmer sofistikerade algoritmer kan införliva realistisk fysik och beräkna bindningsenergierna mellan olika små molekyler och proteiner. Sådana metoder kan förbruka betydande beräkningsresurser, dock, för att uppnå den noggrannhet som behövs. Branschnyttiga simuleringar måste också ge snabba svar. På grund av dragkampen mellan noggrannhet och hastighet, forskare innoverar ständigt, utveckla effektivare algoritmer och förbättra prestanda, Säger Jha.
Detta problem kräver också att hantera beräkningsresurser annorlunda än för många andra storskaliga problem. Istället för att designa en enda simulering som skalar för att använda en hel superdator, forskare kör samtidigt många mindre modeller som formar varandra och banan för framtida beräkningar, en strategi som kallas ensemble-based computing, eller komplexa arbetsflöden.
"Tänk på det här som att försöka utforska ett mycket stort öppet landskap för att försöka hitta var du kanske kan få den bästa läkemedelskandidaten, "Säger Jha. Tidigare har forskare har bett datorer att navigera i detta landskap genom att göra slumpmässiga statistiska val. Vid ett beslut, hälften av beräkningarna kan följa en väg, den andra halvan en annan.
Jha och hans team söker sätt att hjälpa dessa simuleringar att lära av landskapet istället. Att ta in och sedan dela realtidsdata är inte lätt, Jha säger, "och det var det som krävde en del av den tekniska innovationen att göra i stor skala." Han och hans Rutgers-baserade team samarbetar med Peter Coveneys grupp vid University College London om detta arbete.
För att testa denna idé, de har använt algoritmer som förutsäger bindningsaffinitet och har infört strömlinjeformade versioner i ett HTBAC -ramverk, för bindningsaffinitetskalkylator med hög genomströmning. En sådan räknare, känd som ESMACS, hjälper dem att eliminera molekyler som binder dåligt till ett målprotein. Den andra, BANDAR, är mer exakt men mer begränsad i omfattning och kräver 2,5 gånger fler beräkningsresurser. Ändå, det kan hjälpa forskarna att optimera en lovande interaktion mellan ett läkemedel och ett protein. HTBAC -ramverket hjälper dem att implementera dessa algoritmer effektivt, spara den mer intensiva algoritmen för situationer där den behövs.
Teamet demonstrerade idén genom att undersöka 16 läkemedelskandidater från ett molekylbibliotek på GlaxoSmithKline (GSK) med sitt mål, BRD4-BD1-ett protein som är viktigt vid bröstcancer och inflammatoriska sjukdomar. Läkemedelskandidaterna hade samma kärnstruktur men skilde sig åt vid fyra olika områden runt molekylens kanter.
I denna första studie körde teamet tusentals processer samtidigt på 32, 000 kärnor på Blue Waters, en superdator från National Science Foundation (NSF) vid University of Illinois i Urbana-Champaign. De har kört liknande beräkningar på Titan, superdator Cray XK7 vid Oak Ridge Leadership Computing Facility, en användaranläggning för DOE Office of Science. Teamet skilde framgångsrikt mellan bindningen av dessa 16 läkemedelskandidater, den största sådan simulering hittills. "Vi nådde inte bara en oöverträffad skala, "Säger Jha." Vår metod visar förmågan att differentiera. "
De vann sin SCALE -utmärkelse för detta första bevis på konceptet. Utmaningen nu, Jha säger, ser till att det inte bara fungerar för BRD4 utan även för andra kombinationer av läkemedelsmolekyler och proteinmål.
Om forskarna kan fortsätta att utöka sitt tillvägagångssätt, sådana tekniker kan så småningom hjälpa till att påskynda läkemedelsupptäckten och möjliggöra personlig medicin. Men för att undersöka mer realistiska problem, de behöver mer beräkningskraft. "Vi är mitt i denna spänning mellan ett mycket stort kemiskt utrymme som vi, i princip, behöver utforska, och, tyvärr begränsade datorresurser. "säger Jha.
Även när superdatorn expanderar mot exascale, beräkningsforskare kan mer än fylla luckan genom att lägga till mer realistisk fysik till sina modeller. Under överskådlig framtid, forskare måste vara resursstarka för att skala upp dessa beräkningar. Nödvändigheten är innovationsmor, Jha säger, just för att molekylär vetenskap inte kommer att ha den ideala mängden beräkningsresurser för att utföra simuleringar.
Men exascale computing kan hjälpa dem att närma sig sina mål. Förutom att arbeta med University College London och GSK, Jha och hans kollegor samarbetar med Rick Stevens från Argonne National Laboratory och CANcer Distributed Learning Environment (CANDLE) -teamet. Detta samdesignprojekt inom DOE:s Exascale Computing Project bygger djupa neurala nätverk och allmänna maskininlärningstekniker för att studera cancer. Algoritmerna och programvaran inom HTBAC kan komplettera CANDLEs fokus på dessa tillvägagångssätt.
Detta bredare samarbete mellan Jhas grupp, teamet CANDLE och John Chodera's Lab vid Memorial Sloan-Kettering Cancer Center har lett till projektet Integrated and Scalable Prediction of Resistance (INSPIRE). Detta team har redan kört simuleringar på DOE:s Summit -superdator vid Oak Ridge National Laboratory. Det kommer snart att fortsätta detta arbete med Frontera - NSF:s ledarmaskin vid University of Texas i Austin i Texas Advanced Computing Center.
"Vi är sugna på större framsteg och större metodiska förbättringar, "Säger Jha." Vi skulle vilja se hur dessa ganska kompletterande tillvägagångssätt integrativt kan fungera mot denna stora vision. "