Resultat från en analys av artificiella neurala nätverk (ANN) kanske inte är tillförlitliga för molekyler som är alltför olika de som ANN tränades på. De svarta molnen som visas här täcker övergångsmetallkomplex i datamängden vars numeriska representationer är för avlägsna från träningskomplexen för att anses tillförlitliga. Kredit:Massachusetts Institute of Technology
På senare år har maskininlärning har visat sig vara ett värdefullt verktyg för att identifiera nya material med egenskaper optimerade för specifika tillämpningar. Jobbar med stora, väldefinierade datamängder, datorer lär sig att utföra en analysuppgift för att generera ett korrekt svar och sedan använda samma teknik på en okänd datamängd.
Även om detta tillvägagångssätt har väglett utvecklingen av värdefulla nya material, de har främst varit organiska föreningar, noterar Heather Kulik Ph.D. '09, en biträdande professor i kemiteknik. Kulik fokuserar istället på oorganiska föreningar – i synnerhet, de som är baserade på övergångsmetaller, en familj av grundämnen (inklusive järn och koppar) som har unika och användbara egenskaper. I dessa föreningar - kända som övergångsmetallkomplex - förekommer metallatomen i centrum med kemiskt bundna armar, eller ligander, gjord av kol, väte, kväve, eller syreatomer som strålar utåt.
Övergångsmetallkomplex spelar redan viktiga roller inom områden som sträcker sig från energilagring till katalys för tillverkning av finkemikalier – till exempel, för läkemedel. Men Kulik tror att maskininlärning kan utöka användningen ytterligare. Verkligen, hennes grupp har inte bara arbetat med att tillämpa maskininlärning på oorganiska ämnen – ett nytt och utmanande företag – utan också med att använda tekniken för att utforska nytt territorium. "Vi var intresserade av att förstå hur långt vi kunde driva våra modeller för att göra upptäckter - att göra förutsägelser om föreningar som inte har setts tidigare, säger Kulik.
Sensorer och datorer
De senaste fyra åren, Kulik och Jon Paul Janet, en doktorand i kemiteknik, har fokuserat på övergångsmetallkomplex med "spin" - en kvantmekanisk egenskap hos elektroner. Vanligtvis, elektroner förekommer i par, en med snurr upp och den andra med spin ner, så de tar bort varandra och det finns inget nettosnurr. Men i en övergångsmetall, elektroner kan vara oparade, och det resulterande nettospinnet är egenskapen som gör oorganiska komplex av intresse, säger Kulik. "Att skräddarsy hur oparade elektronerna är ger oss en unik ratt för att skräddarsy egenskaper."
Ett givet komplex har ett föredraget spinntillstånd. Men lägg till lite energi – säg, från ljus eller värme – och den kan vända till det andra tillståndet. I processen, det kan uppvisa förändringar i makroskalaegenskaper som storlek eller färg. När energin som behövs för att orsaka vändningen - kallad spin-splittringsenergin - är nära noll, komplexet är en bra kandidat för användning som sensor, eller kanske som en grundläggande komponent i en kvantdator.
Kemister känner till många metall-ligand-kombinationer med spin-splittande energier nära noll, vilket gör dem till potentiella "spin-crossover" (SCO)-komplex för sådana praktiska tillämpningar. Men alla möjligheter är enorma. Spin-splittringsenergin för ett övergångsmetallkomplex bestäms av vilka ligander som kombineras med en given metall, och det finns nästan oändliga ligander att välja mellan. Utmaningen är att hitta nya kombinationer med den önskade egenskapen för att bli SCOs – utan att ta till miljontals trial-and-error-tester i ett labb.
Översätta molekyler till tal
Det vanliga sättet att analysera den elektroniska strukturen hos molekyler är att använda en beräkningsmodelleringsmetod som kallas densitetsfunktionsteori, eller DFT. Resultaten av en DFT-beräkning är ganska exakta - särskilt för organiska system - men att utföra en beräkning för en enskild förening kan ta timmar, eller till och med dagar. I kontrast, ett maskininlärningsverktyg som kallas ett artificiellt neuralt nätverk (ANN) kan tränas att utföra samma analys och sedan göra det på bara några sekunder. Som ett resultat, ANN:er är mycket mer praktiska för att leta efter möjliga SCO:er i det enorma utrymmet av genomförbara komplex.

Den här grafiken representerar ett prov övergångsmetallkomplex. Ett övergångsmetallkomplex består av en central övergångsmetallatom (orange) omgiven av en rad kemiskt bundna organiska molekyler i strukturer som kallas ligander. Kredit:Massachusetts Institute of Technology
Eftersom en ANN kräver en numerisk ingång för att fungera, forskarnas första utmaning var att hitta ett sätt att representera ett givet övergångsmetallkomplex som en serie tal, var och en beskriver en vald egenskap. Det finns regler för att definiera representationer för organiska molekyler, där en molekyls fysiska struktur säger mycket om dess egenskaper och beteende. Men när forskarna följde dessa regler för övergångsmetallkomplex, det fungerade inte. "Den metall-organiska bindningen är väldigt svår att få rätt, " säger Kulik. "Det finns unika egenskaper hos bindningen som är mer varierande. Det finns många fler sätt som elektronerna kan välja att bilda en bindning på." Så forskarna behövde ta fram nya regler för att definiera en representation som skulle vara prediktiv i oorganisk kemi.
Med hjälp av maskininlärning, de utforskade olika sätt att representera ett övergångsmetallkomplex för att analysera spin-splitting energi. Resultaten var bäst när representationen gav mest betoning på egenskaperna hos metallcentrum och metall-ligand-kopplingen och mindre betoning på egenskaperna hos ligander längre ut. Intressant, deras studier visade att representationer som gav mer lika betoning överlag fungerade bäst när målet var att förutsäga andra egenskaper, såsom ligand-metallbindningslängden eller tendensen att acceptera elektroner.
Testar ANN
Som ett test på deras inställning, Kulik och Janet – assisterad av Lydia Chan, en sommarpraktikant från Troy High School i Fullerton, Kalifornien - definierade en uppsättning övergångsmetallkomplex baserade på fyra övergångsmetaller - krom, mangan, järn, och kobolt - i två oxidationstillstånd med 16 ligander (varje molekyl kan ha upp till två). Genom att kombinera dessa byggstenar, de skapade ett "sökutrymme" på 5, 600 komplex - några av dem välbekanta och välstuderade, och några av dem helt okända.
I tidigare arbeten, forskarna hade tränat en ANN på tusentals föreningar som var välkända inom övergångsmetallkemi. För att testa den utbildade ANN:s förmåga att utforska ett nytt kemiskt utrymme för att hitta föreningar med de riktade egenskaperna, de försökte applicera det på poolen av 5, 600 komplex, 113 av vilka den hade sett i den tidigare studien.
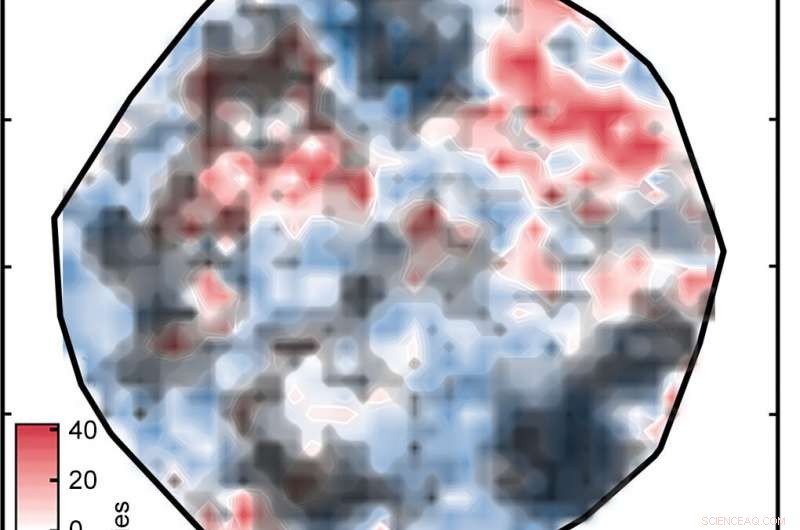
Resultatet var handlingen märkt "Figur 1" i bildspelet ovan, som sorterar komplexen på en yta som bestäms av ANN. De vita områdena indikerar komplex med spin-splittande energier inom 5 kilokalorier per mol noll, vilket innebär att de är potentiellt bra SCO-kandidater. De röda och blå områdena representerar komplex med spin-splittande energier för stora för att vara användbara. De gröna diamanterna som visas i insättningen visar komplex som har järncentra och liknande ligander – med andra ord, relaterade föreningar vars spin-crossover-energier bör vara liknande. Deras utseende i samma område av handlingen är bevis på den goda överensstämmelsen mellan forskarnas representation och komplexets nyckelegenskaper.
Men det finns en hake:inte alla förutsägelser om spinndelning är korrekta. Om ett komplex skiljer sig mycket från de som nätverket tränades på, ANN-analysen kanske inte är tillförlitlig – ett standardproblem när man tillämpar maskininlärningsmodeller på upptäckter inom materialvetenskap eller kemi, konstaterar Kulik. Genom att använda ett tillvägagångssätt som såg framgångsrikt ut i deras tidigare arbete, forskarna jämförde de numeriska representationerna för tränings- och testkomplexen och uteslöt alla testkomplex där skillnaden var för stor.
Fokusera på de bästa alternativen
Utför ANN-analysen av alla 5, 600 komplex tog bara en timme. Men i den verkliga världen, antalet komplex som ska utforskas kan vara tusentals gånger större – och alla lovande kandidater skulle kräva en fullständig DFT-beräkning. Forskarna behövde därför en metod för att utvärdera en stor datamängd för att identifiera eventuella oacceptabla kandidater redan innan ANN-analysen. För detta ändamål, de utvecklade en genetisk algoritm – ett tillvägagångssätt inspirerat av naturligt urval – för att poängsätta individuella komplex och kasta bort de som anses vara olämpliga.
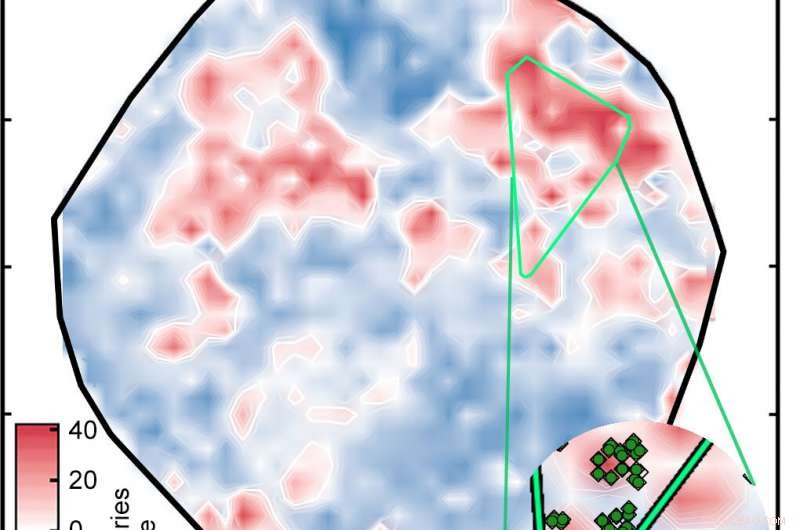
Ett artificiellt neuralt nätverk som tidigare tränats på välkända föreningar analyserades 5, 600 övergångsmetallkomplex för att identifiera potentiella spin-crossover-komplex. Resultatet blev denna handling, där komplex färgas baserat på sin spin-splittringsenergi i kilokalorier per mol (kcal/mol). I lovande kandidater, att energin ligger inom 5 kcal/mol från noll. De ljusgröna diamanterna i insättningen är relaterade komplex. Kredit:Massachusetts Institute of Technology
För att förhandsgranska en datamängd, den genetiska algoritmen väljer först slumpmässigt ut 20 prover från hela uppsättningen av komplex. Den tilldelar sedan ett "fitness"-poäng till varje prov baserat på tre mått. Först, är dess spin-crossover-energi tillräckligt låg för att den ska vara en bra SCO? Att få reda på, det neurala nätverket utvärderar vart och ett av de 20 komplexen. Andra, är komplexet för långt borta från träningsdata? Om så är fallet, spin-crossover-energin från ANN kan vara felaktig. Och slutligen, ligger komplexet för nära träningsdatan? Om så är fallet, forskarna har redan gjort en DFT-beräkning på en liknande molekyl, så kandidaten är inte av intresse i jakten på nya alternativ.
Baserat på sin tredelade utvärdering av de första 20 kandidaterna, den genetiska algoritmen kastar ut olämpliga alternativ och sparar de starkaste till nästa omgång. För att säkerställa mångfalden av de sparade föreningarna, Algoritmen kräver att några av dem muterar lite. Ett komplex kan tilldelas en ny, slumpmässigt utvald ligand, eller två lovande komplex kan byta ligander. Trots allt, om ett komplex ser bra ut, då kan något mycket liknande vara ännu bättre — och målet här är att hitta nya kandidater. Den genetiska algoritmen lägger sedan till några nya, slumpmässigt valda komplex för att fylla i den andra gruppen om 20 och utföra sin nästa analys. Genom att upprepa denna process totalt 21 gånger, den producerar 21 generationer av alternativ. Den fortsätter alltså genom sökutrymmet, låta de starkaste kandidaterna överleva och fortplanta sig, och de olämpliga att dö ut.
Genom att utföra 21-generationsanalysen på hela 5, 600-komplex datauppsättning krävs drygt fem minuter på en vanlig stationär dator, och det gav 372 leads med en bra kombination av hög mångfald och acceptabelt självförtroende. Forskarna använde sedan DFT för att undersöka 56 komplex slumpmässigt valda bland dessa leads, och resultaten bekräftade att två tredjedelar av dem kunde vara bra SCO:er.
Även om en framgångsfrekvens på två tredjedelar kanske inte låter bra, forskarna gör två poänger. Först, deras definition av vad som kan göra en bra SCO var mycket restriktiv:För att ett komplex ska överleva, dess spin-splittringsenergi måste vara extremt liten. Och för det andra, ges ett utrymme på 5, 600 komplex och inget att gå på, hur många DFT-analyser skulle krävas för att hitta 37 leads? Som Janet noterar, "Det spelar ingen roll hur många vi utvärderade med det neurala nätverket eftersom det är så billigt. Det är DFT-beräkningarna som tar tid."
Bäst av alla, genom att använda deras tillvägagångssätt gjorde det möjligt för forskarna att hitta några okonventionella SCO-kandidater som man inte skulle ha tänkt på baserat på vad som har studerats tidigare. "Det finns regler som människor har - heuristik i sina huvuden - för hur de skulle bygga ett spin-crossover-komplex, " säger Kulik. "Vi visade att du kan hitta oväntade kombinationer av metaller och ligander som normalt inte studeras men som kan vara lovande som spin-crossover-kandidater."
Dela de nya verktygen
För att stödja det världsomspännande sökandet efter nytt material, forskarna har införlivat den genetiska algoritmen och ANN i "molSimplify, "gruppen är online, programvara med öppen källkod som alla kan ladda ner och använda för att bygga och simulera övergångsmetallkomplex. För att hjälpa potentiella användare, webbplatsen tillhandahåller handledningar som visar hur man använder nyckelfunktionerna i programvarukoderna med öppen källkod. Utvecklingen av molSimplify började med finansiering från MIT Energy Initiative 2014, och alla elever i Kuliks grupp har bidragit till det sedan dess.
Forskarna fortsätter att förbättra sitt neurala nätverk för att undersöka potentiella SCO:er och för att lägga upp uppdaterade versioner av molSimplify. Under tiden, andra i Kuliks labb utvecklar verktyg som kan identifiera lovande föreningar för andra tillämpningar. Till exempel, ett viktigt fokusområde är katalysatordesign. Doktorand i kemi Aditya Nandy fokuserar på att hitta en bättre katalysator för att omvandla metangas till ett lättare att hantera flytande bränsle som metanol - ett särskilt utmanande problem. "Nu har vi en extern molekyl som kommer in, och vårt komplex – katalysatorn – måste agera på den molekylen för att utföra en kemisk omvandling som äger rum i en hel serie steg, " säger Nandy. "Maskininlärning kommer att vara superanvändbart för att ta reda på de viktiga designparametrarna för ett övergångsmetallkomplex som kommer att göra varje steg i den processen energiskt gynnsamt."
Den här historien återpubliceras med tillstånd av MIT News (web.mit.edu/newsoffice/), en populär webbplats som täcker nyheter om MIT-forskning, innovation och undervisning.