Kredit:Frederik Sandfort/ Pixabay
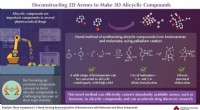
En vardag utan artificiell intelligens är knappt tänkbar i dagens värld. Oräkneliga applikationer inom områden som autonom körning, främmande språk översättning eller medicinsk diagnostik har hittat sin väg in i våra liv. Inom kemisk forskning, för, stora ansträngningar görs för att tillämpa artificiell intelligens (AI), även känd som maskininlärning, effektivt. Dessa teknologier har redan använts för att förutsäga egenskaperna hos enskilda molekyler, vilket gör det lättare för forskare att välja den förening som ska produceras.
Denna produktion, känd som syntes, vanligtvis innebär stora ansträngningar eftersom det finns många möjliga syntesvägar för att producera en målmolekyl. Eftersom framgången för varje enskild reaktion beror på många parametrar, det är inte alltid möjligt, även för erfarna kemister, att förutsäga om en reaktion kommer att äga rum – och ännu mindre hur väl den kommer att fungera. För att råda bot på denna situation, ett team av kemister och datavetare från universitetet i Münster (Tyskland) har gått samman och utvecklat ett AI -verktyg som nu har publicerats i tidskriften Chem .
Bakgrund och metod:
"En kemisk reaktion är ett mycket komplext system, " förklarar Frederik Sandfort, Ph.D. student vid Institutet för organisk kemi och en av publikationens huvudförfattare. "I motsats till förutsägelsen av egenskaper hos enskilda föreningar, en reaktion är samspelet mellan många molekyler och därmed ett flerdimensionellt problem, " tillägger han. Dessutom det finns inga klart definierade "spelregler" som, som i fallet med moderna schackdatorer, förenkla utvecklingen av AI-modeller. Av denna anledning, Tidigare tillvägagångssätt för att noggrant förutsäga reaktionsresultat såsom utbyten eller produkter är mestadels baserade på en tidigare förvärvad förståelse av molekylära egenskaper. "Utvecklingen av sådana modeller kräver stor ansträngning. Dessutom har majoriteten av dem är högspecialiserade och kan inte överföras till andra problem, ", tillägger Frederik Sandfort.
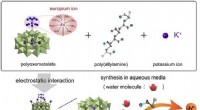
Fokus för det presenterade arbetet låg därför på en allmän tillämplighet av programmet, så att andra kemister lätt kan använda den för sitt eget arbete. För att säkerställa detta, modellen bygger direkt på molekylära strukturer. "Varje organisk förening kan representeras som en graf, i princip som en bild, "förklarar Marius Kühnemund, en annan författare, från området datavetenskap. "På sådana grafer, enkla strukturella frågor - jämförbara med frågan om färger eller former i ett foto - kan göras för att fånga den så kallade kemiska miljön så exakt som möjligt."
Kombinationen av många sådana på varandra följande frågor resulterar i ett så kallat molekylärt fingeravtryck. Dessa enkla nummersekvenser har länge använts inom kemoinformatik för att hitta strukturella likheter och är väl lämpade för datorstödda applikationer. I sitt tillvägagångssätt, författarna använder ett stort antal sådana fingeravtryck för att representera den kemiska strukturen för varje molekyl så exakt som möjligt. "På det här sättet, vi har kunnat utveckla ett robust system som kan användas för att förutsäga helt olika reaktionsresultat, "tillägger Marius Kühnemund, "Samma modell kan användas för att förutsäga både avkastning och stereoselektivitet, vilket är unikt."
Författarna visade att deras program enkelt kan tillämpas och tillåter exakta förutsägelser, speciellt i kombination med modern robotik, genom att använda en datamängd som inte ursprungligen skapades för maskininlärning. "Denna datamängd innehåller endast relativ försäljning av utgångsmaterial och inga exakta avkastningar, " förklarar Frederik Sandfort. "För exakta skördar, kalibreringar måste skapas. Dock, på grund av den höga ansträngningen, detta görs sällan i verkligheten."
Teamet kommer att fortsätta att utveckla sitt program ytterligare och utrusta det med nya funktioner i framtiden. Prof. Frank Glorius är säker:"När det gäller att utvärdera stora mängder komplexa data, datorer är i grunden överlägsna oss. Dock, vårt mål är inte att ersätta syntetiska kemister med maskiner, men för att stödja dem så effektivt som möjligt. Modeller baserade på artificiell intelligens kan avsevärt förändra hur vi närmar oss kemiska synteser. Men vi är fortfarande i början. "