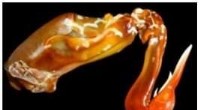
Animation som visar hur coronavirusets spikprotein ändrar form precis innan det binder sig till human cellreceptor. Upphovsman:Illustration tillhandahålls av Mahmoud Moradi.
Beräkningskemisten Mahmoud Moradi kommer att utveckla förbättrad, 3D-simuleringar av molekylär dynamik i coronaviruset spikar glykoproteiner för att få bättre förståelse för hur viruset binder till mänskliga celler.
Kartläggning av hur dessa proteiner genomgår konformationsförändringar för att binda till värdcellsreceptorer är avgörande för utvecklingen av coronavirusvacciner och terapimetoder. Simuleringar är särskilt viktiga eftersom en ram för läkemedelsdesign kommer att kräva dynamisk, tredimensionella visualiseringar av cellstrukturer och beteende, snarare än en statisk bild.
"Som med andra virus, ett avgörande steg i coronavirusinfektionsprocessen är virusinträde, "sa Moradi, biträdande professor vid J. William Fulbright College of Arts and Sciences. "Med coronavirus, vi vet att dessa spikglykoproteiner förmedlar inträde i den mänskliga cellen. Både SARS-CoV-2, orsaken till COVID-19, och SARS-CoV, orsaken till SARS-epidemin 2002-2003, har spikproteiner som fäster till samma receptor i mänskliga celler. "
Moradis arbete är en del av COVID-19 High Performance Computing Consortium, ett samarbete från regeringen, industri och akademiska partners fokuserade på beräkningsresurser för COVID-19-forskning. I spetsen för Vita husets kontor för vetenskap och teknik, det amerikanska energidepartementet och IBM, konsortiets volontärer frigör datortid och resurser på några av världens mest kraftfulla superdatorer.
För att utföra simuleringarna, Moradi har fått tillgång till Frontera, en National Science Foundation-sponsrad superdator inrymd vid University of Texas i Austin. Frontera är den största superdatorn på något universitetscampus.
Moradis projekt gynnas av flera senaste, högupplösta 3D-modeller av spikproteinerna från coronaviruset. Dessa modeller kan användas som initiala strukturer för att påbörja simuleringar som möjliggör analys av proteinernas detaljerade mekanismer och deras beteende vid viral inträde. Förbättrad, detaljerade simuleringar av sådan molekylär dynamik ger en fullständig bild av proteiners strukturella förändringar, liksom hur de binder till angiotensin-konverterande enzym 2, den specifika humana cellreceptorn.
Moradis forskning ligger i skärningspunkten mellan biologi, fysik, kemi, matematik, statistik och datavetenskap. Hans biomolekylära simuleringar och beräkningsteorier förklarar hur proteiner, arbetshästmolekylerna i celler, fungerar på molekylär nivå. Hans arbete förbättrar geometriska modeller för att beskriva hur proteiner ändrar form och hur dessa förändringar påverkar ett proteins beteende. I februari, han fick $ 650, 000 National Science Foundation Faculty Early Career Development award för detta arbete.