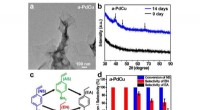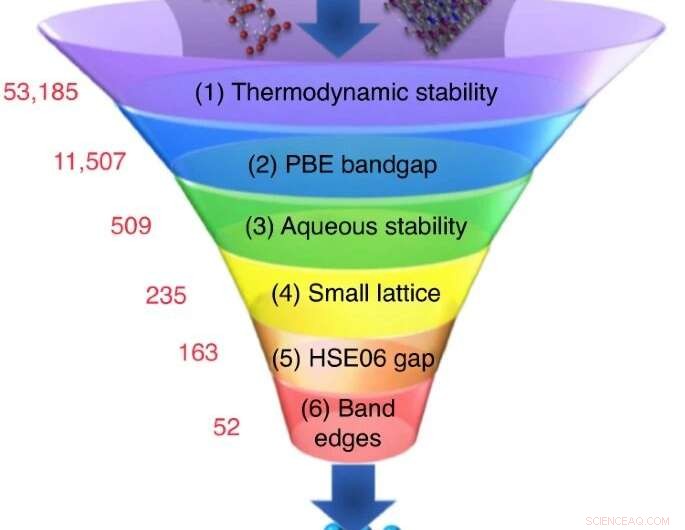
Ett stort antal kandidatmaterial väljs bland experimentella eller beräkningsdatabaser, och en sekvens av screeningberäkningar minskar deras antal till en liten uppsättning kandidater med de mest lovande egenskaperna. Upphovsman:Nicola Marzari
Nicola Marzari, chef för laboratoriet för teori och simulering av material på EFPL och chef för NCCR MARVEL, har just publicerat en översyn av metoder för elektronisk struktur som en del av en specialutgåva Insight on Computational Materials Design, publicerad av Naturmaterial . Artikeln, skriven med Andrea Ferretti från CNR – Instituto Nanoscienze och Chris Wolverton från Northwestern University, ger en översikt över dessa metoder, diskuterar deras tillämpning på förutsägelse av materialegenskaper, och undersöker olika strategier som används för att rikta de bredare målen för materialdesign och upptäckt. Blickar framåt, författarna överväger nya utmaningar i beräkningarnas förutsägbara noggrannhet, och för att hantera materialens och anordningernas komplexitet i verkligheten. De betonar också vikten av beräkningsinfrastrukturer som stöder sådan forskning, och hur planeringen för att finansiera dessa och de stödjande karriärmodellerna bara har börjat dyka upp.
Under de senaste 20 åren har första principer simuleringar har blivit kraftfulla, mycket använda verktyg i många, olika områden inom vetenskap och teknik. Från nanoteknik till planetvetenskap, från metallurgi till kvantmaterial, de har påskyndat identifieringen, karakterisering, och optimering av material enormt. De har lett till häpnadsväckande förutsägelser-från ultrasnabb värmetransport till elektron-fononförmedlad supraledning i hydrider till framväxten av platta band i tvinnat tvåskiktigt grafen-som har inspirerat anmärkningsvärda experiment.
Den nuvarande pushen för att komplettera experiment med simuleringar; fortsatt, snabb tillväxt i datorkapacitetskapacitet; förmågan hos maskininlärning och artificiell intelligens att påskynda materialupptäckten liksom löftet om störande acceleratorer som kvantberäkning för exponentiellt dyra uppgifter innebär att det är uppenbart att dessa metoder kommer att bli allt mer relevanta med tiden. Det är en lämplig tid då att granska möjligheterna såväl som begränsningarna för de elektroniska strukturmetoder som ligger till grund för dessa simuleringar. Marzari, Ferretti och Wolverton tar upp denna uppgift i artikeln "Elektroniska strukturmetoder för materialdesign, "har just publicerats i Naturmaterial .
"Simuleringar misslyckas inte på spektakulära sätt men kan subtilt skifta från att vara ovärderliga till knappt tillräckligt bra till bara värdelösa, "sa författarna i tidningen." Orsakerna till misslyckande är många, från att sträcka ut metodernas möjligheter till att överge komplexiteten hos verkliga material. Men simuleringar är också oersättliga:De kan bedöma material vid tryck- och temperaturförhållanden så extrema att inget experiment på jorden kan replikera, de kan med ständigt ökande smidighet utforska det stora utrymmet av materialfaser och kompositioner i jakten på det svårfångade materialgenombrottet, och de kan direkt identifiera de mikroskopiska orsakerna och ursprunget för en makroskopisk egenskap. Sista, de delar med alla grenar av beräkningsvetenskap ett nyckelelement i forskningen:De kan göras reproducerbara och öppna och delbara på sätt som ingen fysisk infrastruktur någonsin kommer att vara. "
Författarna tittar först på ramen för densitetsfunktionell teori (DFT) och ger en överblick över de allt mer komplexa tillvägagångssätten som kan förbättra noggrannheten eller utöka omfattningen av simuleringar. De diskuterar sedan de möjligheter som beräkningsmaterialvetenskap har utvecklat för att utnyttja denna verktygslåda och leverera förutsägelser för materialens egenskaper under realistiska förhållanden med ständigt ökande komplexitet. Till sist, de belyser hur fysik- eller datadrivna tillvägagångssätt kan ge rationella, hög genomströmning, eller vägar för artificiell intelligens för materialupptäckt, och förklara hur sådana insatser förändrar hela forskningsekosystemet.
Blickar framåt, författarna säger att utveckla metoder som kan bedöma den termodynamiska stabiliteten, syntesförhållanden, tillverkbarhet, och tolerans för de förutsagda egenskaperna för inneboende och yttre defekter i nya material kommer att vara en betydande utmaning. Forskare kan behöva förstärka DFT-uppskattningar med mer avancerade elektroniska strukturmetoder eller maskininlärningsalgoritmer för att förbättra noggrannheten, och använda beräkningsmetoder för att hantera realistiska förhållanden som vibrationsentropier, koncentrationen av defekter och tillämpade elektrokemiska potentialer.
Till sist, med tanke på den utökade roll som sådana metoder sannolikt kommer att spela under de kommande decennierna, författarna noterar att stöd och planering för den nödvändiga beräkningsinfrastrukturen - allmänt använd vetenskaplig programvara, verifiering av koder och validering av teorier, spridning och kurering av beräkningsdata, verktyg och arbetsflöden samt tillhörande karriärmodeller som detta innebär och kräver - har bara börjat dyka upp.