Kredit:Pixabay/CC0 Public Domain
Kemiingenjörer och materialforskare letar ständigt efter nästa revolutionerande material, kemikalier och droger. Ökningen av metoder för maskininlärning påskyndar upptäcktsprocessen, som annars kan ta år. "Helst sett är målet att träna en maskininlärningsmodell på ett fåtal befintliga kemiska prover och sedan låta den producera så många tillverkningsbara molekyler av samma klass som möjligt, med förutsägbara fysikaliska egenskaper", säger Wojciech Matusik, professor i elektroteknik och datavetenskap vid MIT. "Om du har alla dessa komponenter kan du bygga nya molekyler med optimala egenskaper, och du vet också hur man syntetiserar dem. Det är den övergripande visionen som människor i det utrymmet vill uppnå."
Men nuvarande tekniker, främst djupinlärning, kräver omfattande datauppsättningar för träningsmodeller, och många klassspecifika kemiska datauppsättningar innehåller en handfull exempelföreningar, vilket begränsar deras förmåga att generalisera och generera fysiska molekyler som skulle kunna skapas i den verkliga världen.
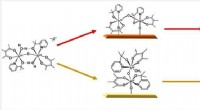
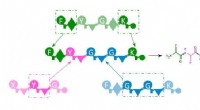
Nu tar ett nytt dokument från forskare vid MIT och IBM detta problem med hjälp av en generativ grafmodell för att bygga nya syntetiserbara molekyler inom samma kemiska klass som deras träningsdata. För att göra detta behandlar de bildningen av atomer och kemiska bindningar som en graf och utvecklar en grafgrammatik – en lingvistisk analogi av system och strukturer för ordordning – som innehåller en sekvens av regler för att bygga molekyler, som monomerer och polymerer. Genom att använda grammatiken och produktionsreglerna som härleddes från utbildningsuppsättningen kan modellen inte bara omvända sina exempel, utan kan skapa nya sammansättningar på ett systematiskt och dataeffektivt sätt. "Vi byggde i princip ett språk för att skapa molekyler", säger Matusik. "Denna grammatik är i grunden den generativa modellen."
Matusiks medförfattare inkluderar MIT-studenter Minghao Guo, som är huvudförfattare, och Beichen Li samt Veronika Thost, Payal Das och Jie Chen, forskningspersonal med IBM Research. Matusik, Thost och Chen är anslutna till MIT-IBM Watson AI Lab. Deras metod, som de har kallat data-effektiv grafgrammatik (DEG), kommer att presenteras på den internationella konferensen om läranderepresentationer.
"Vi vill använda denna grammatikrepresentation för monomer- och polymergenerering, eftersom denna grammatik är förklarlig och uttrycksfull", säger Guo. "Med bara ett fåtal av produktionsreglerna kan vi skapa många typer av strukturer."
En molekylär struktur kan ses som en symbolisk representation i en graf - en sträng av atomer (noder) förenade med kemiska bindningar (kanter). I denna metod låter forskarna modellen ta den kemiska strukturen och kollapsa en understruktur av molekylen ner till en nod; detta kan vara två atomer förbundna med en bindning, en kort sekvens av bundna atomer eller en ring av atomer. Detta görs upprepade gånger och skapar produktionsreglerna allt eftersom, tills en enda nod finns kvar. Reglerna och grammatiken kan sedan tillämpas i omvänd ordning för att återskapa träningsuppsättningen från grunden eller kombineras i olika kombinationer för att producera nya molekyler av samma kemiska klass.
"Befintliga grafgenereringsmetoder skulle producera en nod eller en kant sekventiellt åt gången, men vi tittar på strukturer på högre nivå och, specifikt, utnyttjar kemikunskaper, så att vi inte behandlar de individuella atomerna och bindningarna som enheten. Detta förenklar genereringsprocessen och gör det också mer dataeffektivt att lära sig”, säger Chen.
Vidare optimerade forskarna tekniken så att grammatiken nerifrån och upp var relativt enkel och okomplicerad, så att den tillverkade molekyler som kunde göras.
"Om vi ändrar ordningen för att tillämpa dessa produktionsregler skulle vi få en annan molekyl; dessutom kan vi räkna upp alla möjligheter och generera massor av dem", säger Chen. "Några av dessa molekyler är giltiga och några av dem inte, så inlärningen av själva grammatiken är faktiskt att räkna ut en minimal samling av produktionsregler, så att andelen molekyler som faktiskt kan syntetiseras maximeras." Medan forskarna koncentrerade sig på tre träningsset med mindre än 33 prover vardera - akrylater, kedjeförlängare och isocyanater - noterar de att processen kan tillämpas på vilken kemisk klass som helst.
För att se hur deras metod fungerade testade forskarna DEG mot andra toppmoderna modeller och tekniker, och tittade på procentandelar av kemiskt giltiga och unika molekyler, mångfalden av de som skapades, framgångsgraden för retrosyntes och procentandelen av molekyler som tillhör träningsdatans monomerklass.
"Vi visar tydligt att, för syntetiserbarhet och medlemskap, överträffar vår algoritm alla befintliga metoder med mycket stor marginal, medan den är jämförbar för vissa andra allmänt använda mätvärden", säger Guo. Vidare, "det som är fantastiskt med vår algoritm är att vi bara behöver cirka 0,15 procent av den ursprungliga datamängden för att uppnå mycket liknande resultat jämfört med toppmoderna metoder som tränar på tiotusentals prover. Vår algoritm kan specifikt hantera problemet med datagleshet."
Inom den närmaste framtiden planerar teamet att ta itu med att skala upp denna grammatikinlärningsprocess för att kunna generera stora grafer, samt producera och identifiera kemikalier med önskade egenskaper.
På vägen ser forskarna många tillämpningar för DEG-metoden, eftersom den är anpassningsbar utöver att generera nya kemiska strukturer, påpekar teamet. En graf är en mycket flexibel representation, och många enheter kan symboliseras i denna form - robotar, fordon, byggnader och elektroniska kretsar, till exempel. "I huvudsak är vårt mål att bygga upp vår grammatik, så att vår grafiska representation kan användas i stor utsträckning över många olika domäner", säger Guo, eftersom "DEG kan automatisera designen av nya enheter och strukturer", säger Chen. + Utforska vidare
Denna berättelse är återpublicerad med tillstånd av MIT News (web.mit.edu/newsoffice/), en populär webbplats som täcker nyheter om MIT-forskning, innovation och undervisning.