Föreställ dig att syntetisera och sedan testa över 50 olika komplexa molekyler för att identifiera den mest effektiva katalysatorn för en viss kemisk reaktion. Det traditionella tillvägagångssättet för att utveckla nya katalysatorer för kemiska reaktioner på detta "prova och se"-sätt är ofta extremt arbetsintensivt, vilket kräver många upprepade experiment med potentiella kandidatmolekyler. Den nu allmänt förekommande tekniken för maskininlärning kan göra denna uppgift mycket effektivare genom att förutsäga prestanda hos katalysatorer i förväg baserat på teoretiska egenskaper.
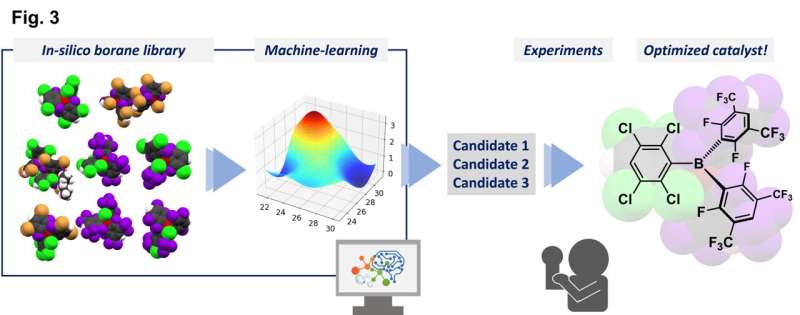
I en studie publicerad i Nature Communications , använde forskare från Osaka University ett datorbibliotek med molekyler som har syntetiserats tillsammans med molekyler som är helt teoretiska för tillfället för att hitta den bästa katalysatorn för en specifik kemisk reaktion.
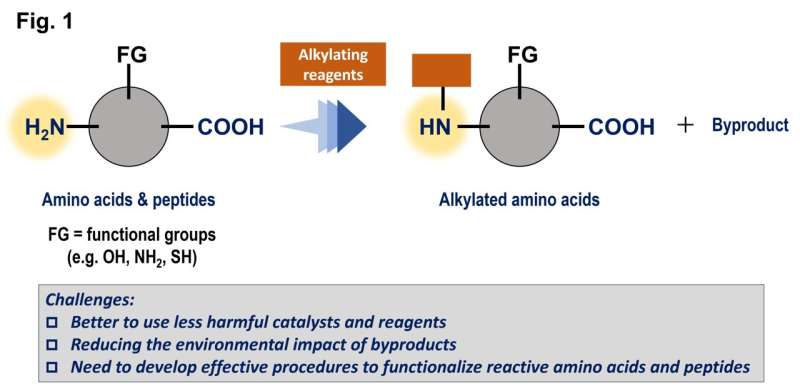
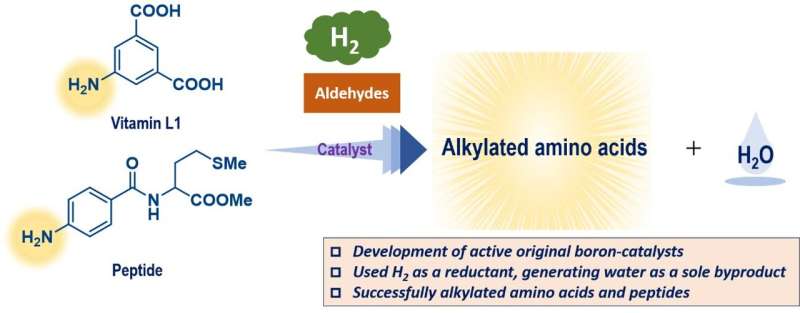
Målet med arbetet var att hitta bättre sätt att lägga till grupper av kol till aminosyror och peptider, som är mycket vanliga i levande organismer, för att modifiera egenskaperna hos dessa föreningar. Liksom många reaktioner förbättras dessa processer av katalysatorer, men en traditionell metallbaserad katalysator är ofta giftig och/eller dyr.
Denna studie syftade till att använda triarylboraner som katalysatorer, men på grund av deras relativt komplexa strukturer finns det potentiellt hundratals möjligheter. Dessa föreningar är baserade på bor, som är ett huvudgruppselement som är relativt billigt och mindre giftigt.
"Utvärderingen av molekylära katalysatorer för organisk syntes kan vara extremt tidskrävande", säger huvudförfattaren till studien Yusei Hisata. "När det gäller triarylboranerna som används i vårt arbete kan många permutationer av molekylära strukturer kräva månaders studier bara för att identifiera den optimala kandidaten."
Forskarna kombinerade experimentella data från ett begränsat antal syntetiserade triarylboraner med egenskaper som förutspåtts för andra molekyler som ännu inte har syntetiserats, med hjälp av teoretiska beräkningar, för att skapa ett bibliotek med 54 möjliga katalysatorer.
"Denna process utvärderade parametrar som vi förutspådde skulle påverka reaktionens framsteg", förklarar Yoichi Hoshimoto, motsvarande författare. "Dessa inkluderade faktorer som den molekylära orbitala energinivån och energibarriärerna för vissa processer."
En Gaussisk processregression med användning av in silico-biblioteket identifierade en lovande kandidat, och tester med denna triarylboran visade en hög prestandanivå. Denna förening kan främja reaktionerna av en aminosyra i mycket höga utbyten och tolerera närvaron av många olika funktionella grupper. Som en extra fördel genererar dessa reaktioner endast vatten som en ofarlig biprodukt eftersom de framgångsrikt använde molekylärt väte, H2 , som ett reagens.
Detta arbete undersökte också andra sätt att minska processens miljöpåverkan och fann att det farliga lösningsmedlet tetrahydrofuran kunde ersättas med det mindre giftiga alternativet 4-metyltetrahydropyran.
Moderna kemister möter ökande krav, och de jonglerar med att utveckla nya synteser med begränsade kamrater samtidigt som de tar hänsyn till miljöpåverkan, effektivitet, kostnad, hållbarhet och andra faktorer. Denna studie visar ett viktigt steg framåt i användningen av maskininlärning för att effektivisera utvecklingen av nya kemiska processer och belyser hur dessa nya processer kan införliva förändringar som samverkar för att generera gröna system.
Mer information: Yusei Hisata et al, In-silico-assisterad derivatisering av triarylboraner för katalytisk reduktiv funktionalisering av anilin-härledda aminosyror och peptider med H2, Nature Communications (2024). DOI:10.1038/s41467-024-47984-0
Journalinformation: Nature Communications
Tillhandahålls av Osaka University