Forskare vid UC San Diego har utvecklat en maskininlärningsalgoritm för att simulera den tidskrävande kemin som är involverad i de tidigaste faserna av läkemedelsupptäckten, vilket avsevärt skulle kunna effektivisera processen och öppna dörrar för behandlingar som aldrig tidigare har setts.
Att identifiera läkemedelskandidater för ytterligare optimering involverar vanligtvis tusentals individuella experiment, men den nya plattformen för artificiell intelligens (AI) kan potentiellt ge samma resultat på en bråkdel av tiden. Forskarna använde det nya verktyget, som beskrivs i Nature Communications , för att syntetisera 32 nya läkemedelskandidater för cancer.
Tekniken är en del av en ny men växande trend inom läkemedelsvetenskapen att använda AI för att förbättra upptäckt och utveckling av läkemedel.
"För några år sedan var AI ett smutsigt ord i läkemedelsindustrin, men nu är trenden definitivt den motsatta, där bioteknikföretag har svårt att samla in pengar utan att ta upp AI i sin affärsplan", säger seniorförfattaren Trey Ideker, professor vid Institutionen för medicin vid UC San Diego School of Medicine och adjungerad professor i bioteknik och datavetenskap vid UC San Diego Jacobs School of Engineering.
"AI-styrd läkemedelsupptäckt har blivit ett mycket aktivt område inom industrin, men till skillnad från de metoder som utvecklas i företag, gör vi vår teknik öppen källkod och tillgänglig för alla som vill använda den."
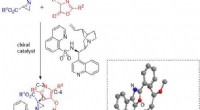
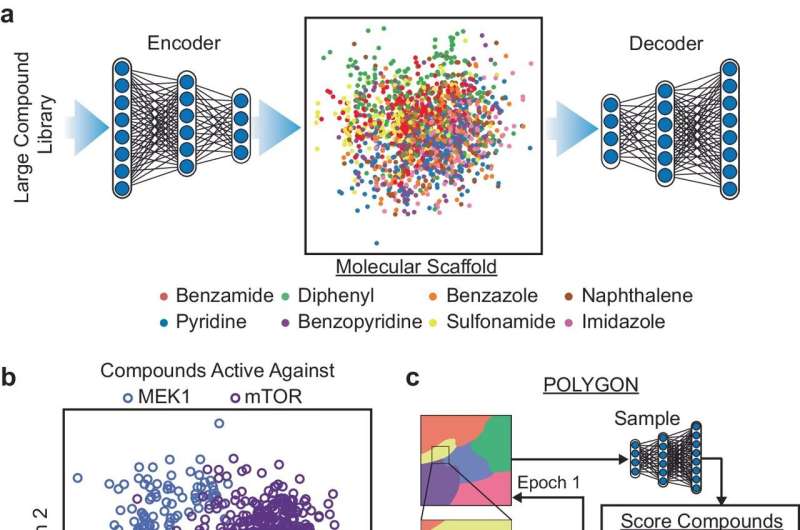
Den nya plattformen, kallad POLYGON, är unik bland AI-verktyg för läkemedelsupptäckt genom att den kan identifiera molekyler med flera mål, medan befintliga läkemedelsupptäcktsprotokoll för närvarande prioriterar enstaka målterapier. Läkemedel med flera mål är av stort intresse för läkare och forskare på grund av deras potential att ge samma fördelar som kombinationsterapi, där flera olika läkemedel används tillsammans för att behandla cancer, men med färre biverkningar.
"Det tar många år och miljontals dollar att hitta och utveckla ett nytt läkemedel, speciellt om vi pratar om ett med flera mål", säger Ideker. "De sällsynta få multi-target läkemedel vi har upptäcktes till stor del av en slump, men den här nya tekniken kan hjälpa till att ta chansen ur ekvationen och kickstarta en ny generation av precisionsmedicin."
Forskarna tränade POLYGON på en databas med över en miljon kända bioaktiva molekyler som innehåller detaljerad information om deras kemiska egenskaper och kända interaktioner med proteinmål. Genom att lära sig av mönster som finns i databasen kan POLYGON skapa ursprungliga kemiska formler för nya läkemedelskandidater som sannolikt har vissa egenskaper, såsom förmågan att hämma specifika proteiner.
"Precis som AI nu är väldigt bra på att skapa originalritningar och bilder, som att skapa bilder av mänskliga ansikten baserat på önskade egenskaper som ålder eller kön, kan POLYGON generera ursprungliga molekylära föreningar baserade på önskade kemiska egenskaper," sa Ideker.
"I det här fallet, istället för att tala om för AI hur gammalt vi vill att vårt ansikte ska se ut, berättar vi hur vi vill att vårt framtida läkemedel ska interagera med sjukdomsproteiner."
För att sätta POLYGON på prov använde forskarna det för att generera hundratals läkemedelskandidater som riktar sig mot olika par av cancerrelaterade proteiner.
Av dessa syntetiserade forskarna 32 molekyler som hade de starkast förutsagda interaktionerna med MEK1- och mTOR-proteinerna, ett par cellulära signalproteiner som är ett lovande mål för cancerkombinationsterapi. Dessa två proteiner är vad forskare kallar syntetiskt dödliga, vilket betyder att det räcker att hämma båda tillsammans för att döda cancerceller även om det inte är fallet att hämma en ensam.
Forskarna fann att läkemedlen de syntetiserade hade betydande aktivitet mot MEK1 och mTOR, men hade få reaktioner utanför målet med andra proteiner. Detta tyder på att ett eller flera av de läkemedel som identifierats av POLYGON skulle kunna rikta in sig på båda proteinerna som en cancerbehandling, vilket ger en lista över val för finjustering av mänskliga kemister.
"När du väl har fått läkemedelskandidaterna behöver du fortfarande göra all annan kemi som krävs för att förfina dessa alternativ till en enda effektiv behandling", sa Ideker. "Vi kan och bör inte försöka eliminera mänsklig expertis från läkemedelsupptäcktspipelinen, men vad vi kan göra är att förkorta några steg i processen."
Trots denna försiktighet är forskarna optimistiska att AI:s möjligheter för läkemedelsupptäckt bara undersöks.
"Att se hur det här konceptet kommer att utspela sig under det kommande decenniet, både inom den akademiska världen och i den privata sektorn, kommer att bli väldigt spännande", säger Ideker. "Möjligheterna är praktiskt taget oändliga."
Mer information: Brenton P. Munson et al, De novo generation av multi-target föreningar med djup generativ kemi, Nature Communications (2024). DOI:10.1038/s41467-024-47120-y. www.nature.com/articles/s41467-024-47120-y
Journalinformation: Nature Communications
Tillhandahålls av University of California - San Diego