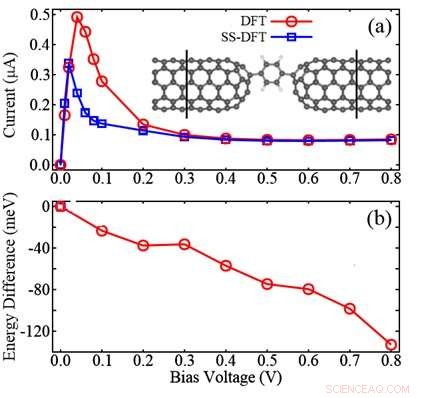
Figur visar jämförelsen mellan SS-DFT och allmänt använda DFT-metoder för en molekylär anordning bestående av två kolnanorör (CNT) -elektroder och en bensenmolekyl däremellan:(a) beräknade IV-kurvor; (b) energidifferensen beräknad genom att subtrahera DFT-energin från SS-DFT-energin. Figuren visar att SS-DFT förutspår det energiskt mer stabila transporttillståndet med lägre elektriska strömmar jämfört med den DFT-baserade metoden. Upphovsman:Zhang Chun
NUS beräkningsforskare har utvecklat en ny version av densitetsfunktionell teori (DFT) för att studera nanoskalaenheter.
Elektroniska enheter blir mindre och har större funktionalitet. Detta görs möjligt genom att minska storleken på de elektroniska komponenterna. När deras storlek minskar, egenskaperna hos dessa molekylära enheter blir mycket mer känsliga för deras yttre miljö. Beräkningsmetoder krävs för att simulera och förutsäga egenskaperna hos sådana små enheter. En av dem är densitetsfunktionell teori. Dessa metoder är utvecklade utifrån första principer, bestående av grundläggande och grundläggande kunskap som vi redan känner till. NUS beräkningsforskare har förfinat teorin för att ta hänsyn till icke-jämviktseffekter som förekommer under driften av enheterna (t.ex. när ett batteri är anslutet till ett kvantsystem). Detta leder till en mer exakt och exakt förutsägelse.
Prof ZHANG Chun och hans doktorand studerande, LIU Shuanglong tillsammans med forskare, Dr Argo NURBAWONO, från Institutionen för fysik, NUS har utvecklat en mer allmän version av den populära och allmänt använda densitetsfunktionsteorin (DFT) som kan tillämpas på stationära icke-jämviktssituationer. De införde en ytterligare grad av frihet, känd som icke-jämvikt elektrontäthet, in i modelleringen med de första principerna. Detta tar hänsyn till de bias-inducerade icke-jämviktseffekterna när en molekylär enhet fungerar under en begränsad förspänning. Denna nya version av teorin är känd som steady-state DFT (SS-DFT).
Forskarna har visat att den mycket använda DFT i princip inte är korrekt i ett steady-state-jämviktsscenario. I en sådan situation, två olika parametrar, den totala elektrontätheten och densiteten hos strömbärande elektroner, behövs för att bestämma egenskaperna hos det motsvarande icke-jämviktssystemet. Den nya teorin har implementerats i det spanska initiativet för elektroniska simuleringar med tusentals atomer (SIESTA) beräkningspaket för att studera elektroniska/ transportegenskaper hos olika enheter i molekylskala.
SS-DFT ger ett pålitligt teoretiskt verktyg för att förstå och framtida design av nya molekylära enheter med förbättrad funktionalitet. Det SS-DFT-baserade beräkningspaketet används nu av många forskargrupper över hela världen. Den används för att förklara spännande transportfenomen som observerats experimentellt på molekylär nivå och för att utforma nya typer av molekylära enheter.
Forskarna planerar att utöka teorins tillämplighet genom att inkludera andra fysiska effekter, såsom elektron-fonon-interaktioner och spin-orbital-koppling. De avser också att förbättra beräkningseffektiviteten så att den kan användas för att modellera stora system runt 1, 000 atomer.